Objective: To describe the long-term safety, tolerability, and symptom trajectory with the long-acting injectable antipsychotic aripiprazole lauroxil (AL) in patients with DSM-5-diagnosed schizophrenia followed for up to 180 weeks (3.5 years).
Methods: Long-term safety of 2 fixed doses of AL (441 or 882 mg every 4 weeks) was assessed during up to 180 weeks (3.5 years) of continuous AL exposure using data from 2 sequential long-term safety studies. Safety metrics included adverse events (AEs), AEs leading to study discontinuations, physical examinations, laboratory parameters, and extrapyramidal symptom (EPS) rating scales. Symptom trajectory was assessed in post hoc analyses using Positive and Negative Syndrome Scale total (PANSST) and Clinical Global Impressions-Severity of Illness scale (CGI-S) scores.
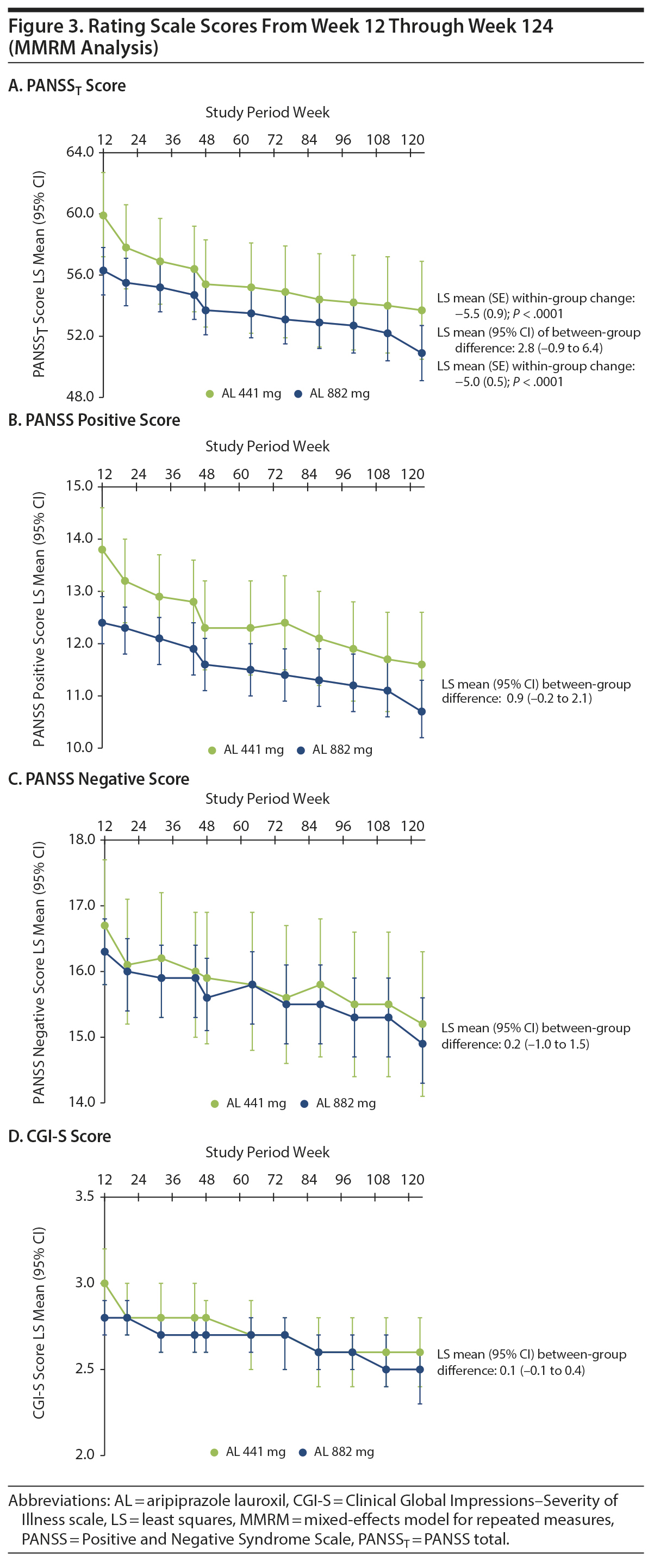
Results: A total of 478 patients entered the 52-week study and were included in the safety analysis. After the first 52 weeks, safety assessments revealed no new safety concerns and were consistent with the known safety profile of aripiprazole. AEs were reported by 57.5% of patients (441 mg, 52.7%; 882 mg, 59.0%). EPS-related AEs occurred in 12.8% of patients (441 mg, 9.1%; 882 mg, 13.9%). In the post hoc analysis (n = 432), least-squares mean (SE) PANSST scores improved significantly from weeks 12 to 124 with AL 441 mg (−5.5 [0.9]) and 882 mg (−5.0 [0.5]; both P < .0001). CGI-S scores followed a similar pattern of improvement.
Conclusions: The AL safety profile over 180 weeks (3.5 years) of follow-up was consistent with prior 52-week results. Continued therapeutic efficacy, based on PANSST and CGI-S scores, was observed throughout the post hoc analysis period.
Trial Registration: ClinicalTrials.gov identifier: NCT01626456; ClinicalTrials.gov identifier: NCT01895452
Beyond 52-Week Long-Term Safety:
Long-Term Outcomes of Aripiprazole Lauroxil for Patients With Schizophrenia Continuing in an Extension Study
ABSTRACT
Objective: To describe the long-term safety, tolerability, and symptom trajectory with the long-acting injectable antipsychotic aripiprazole lauroxil (AL) in patients with DSM-5-diagnosed schizophrenia followed for up to 180 weeks (3.5 years).
Methods: Long-term safety of 2 fixed doses of AL (441 or 882 mg every 4 weeks) was assessed during up to 180 weeks (3.5 years) of continuous AL exposure using data from 2 sequential long-term safety studies. Safety metrics included adverse events (AEs), AEs leading to study discontinuations, physical examinations, laboratory parameters, and extrapyramidal symptom (EPS) rating scales. Symptom trajectory was assessed in post hoc analyses using Positive and Negative Syndrome Scale total (PANSST) and Clinical Global Impressions-Severity of Illness scale (CGI-S) scores.
Results: A total of 478 patients entered the 52-week study and were included in the safety analysis. After the first 52 weeks, safety assessments revealed no new safety concerns and were consistent with the known safety profile of aripiprazole. AEs were reported by 57.5% of patients (441 mg, 52.7%; 882 mg, 59.0%). EPS-related AEs occurred in 12.8% of patients (441 mg, 9.1%; 882 mg, 13.9%). In the post hoc analysis (n = 432), least-squares mean (SE) PANSST scores improved significantly from weeks 12 to 124 with AL 441 mg (−5.5 [0.9]) and 882 mg (−5.0 [0.5]; both P < .0001). CGI-S scores followed a similar pattern of improvement.
Conclusions: The AL safety profile over 180 weeks (3.5 years) of follow-up was consistent with prior 52-week results. Continued therapeutic efficacy, based on PANSST and CGI-S scores, was observed throughout the post hoc analysis period.
Trial Registration: ClinicalTrials.gov identifiers: NCT01626456; NCT01895452
J Clin Psychiatry 2020;81(5):19m12835
To cite: Lauriello J, Claxton A, Du Y, et al. Beyond 52-week long-term safety: long-term outcomes of aripiprazole lauroxil for patients with schizophrenia continuing in an extension study. J Clin Psychiatry. 2020;81(5):19m12835.
To share: https://doi.org/10.4088/JCP.19m12835
© Copyright 2020 Physicians Postgraduate Press, Inc.
aDepartment of Psychiatry and Human Behavior, Thomas Jefferson University, Philadelphia, Pennsylvania
bAlkermes, Inc, Waltham, Massachusetts
cDr Weiden is currently affiliated with Karuna Therapeutics, Inc, Boston, Massachusetts.
*Corresponding author: John Lauriello, MD, Department of Psychiatry and Human Behavior, Thomas Jefferson University, 833 Chestnut St, Ste 210, Philadelphia, PA 19107 ([email protected]).
Notice of correction 10/2/2020: The second sentence in the fifth paragraph of the Discussion has been corrected to include the words “placebo-adjusted.”
Continuous, uninterrupted, and long-term antipsychotic treatment is widely recognized as a fundamental recommendation in treatment guidelines for schizophrenia.1 Long-term antipsychotic use reduces the risk of relapse, whereas inappropriate dose reductions or gaps in treatment are associated with an increased risk of hospitalization.1,2 Long-acting injectable antipsychotic drugs are a valuable treatment option for improving adherence, reducing treatment gaps, and improving clinical outcomes.3
Aripiprazole lauroxil (AL) is an atypical long-acting antipsychotic for intramuscular injection that was developed as a prodrug of aripiprazole and approved for the treatment of schizophrenia in adults.4 The efficacy, safety, and tolerability of AL 441 mg or 882 mg every 4 weeks for the treatment of schizophrenia were demonstrated in a 12-week phase 3 randomized, double-blind, placebo-controlled trial.5 The adverse event (AE) profile associated with AL in that 12-week study was consistent with that observed with oral aripiprazole at doses indicated for schizophrenia (10-30 mg/d).
A 52-week outpatient study6 evaluating AL safety in patients with stable schizophrenia has been previously reported; the observed safety profile was similar to that noted in the acute, 12-week study.5 Here, we extend 1-year follow-up results with data from a second, contiguous long-term extension (LTE) study that included an additional 1 to 2.5 years of AL exposure. By combining these studies, the current analysis provides aggregate safety and tolerability data and details durability of effect and symptom trajectory associated with up to 180 weeks (3.5 years) of AL treatment in outpatients with schizophrenia.
METHODS
Both the 52-week safety study (ALK9072-003EXT; ClinicalTrials.gov identifier: NCT01626456) and the LTE (ALK9072-003EXT2; ClinicalTrials.gov identifier: NCT01895452) were phase 3 studies designed and carried out in accordance with the principles of Good Clinical Practice that have their origin in the Declaration of Helsinki and its amendments,7 with local regulations, and International Council for Harmonisation guidelines.8 Study protocols and amendments were approved by the independent ethics committee/institutional review board for each study site. All patients provided written informed consent before participating in both studies.
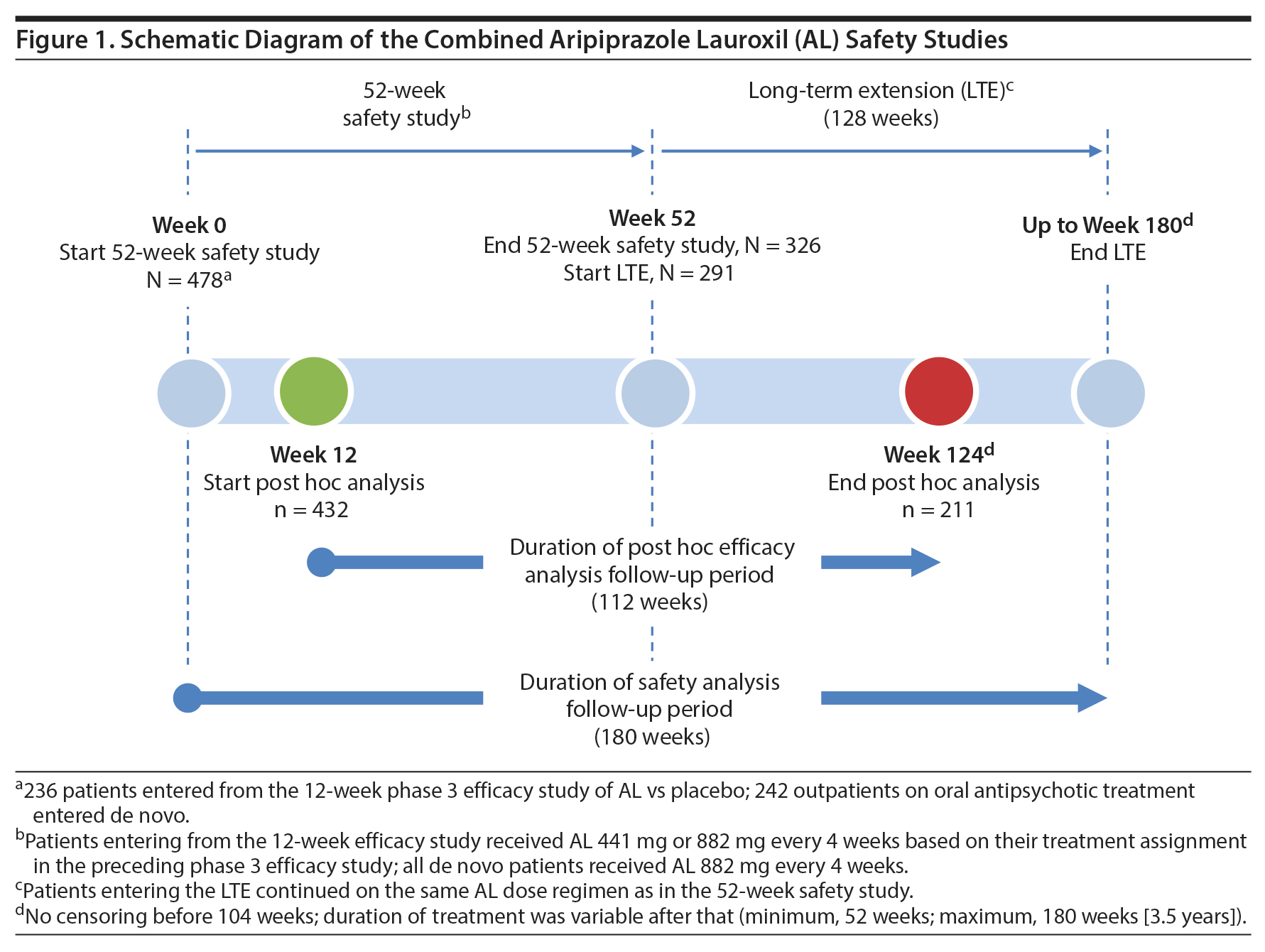
The 52-week study was conducted between June 2012 and April 20156; the continuation LTE was conducted between July 2013 and June 2016. This report includes integrated safety data from the 52-week study along with unpublished data for the additional time in the LTE. When the 52-week and LTE data are combined, the duration of individual patient follow-up extends to 180 weeks (3.5 years) (Figure 1).
Study Design and Treatments
52-week safety study. The initial 52-week safety study was a phase 3 multicenter, open-label investigation in which patients with stable schizophrenia received aripiprazole lauroxil either 441 mg or 882 mg every 4 weeks for a total of up to 13 doses.6
Long-term extension study. After completing the 52-week study, patients could enroll in the open-label LTE and continue their previous AL dosing regimen (Figure 1). Because the LTE had a definitive termination date (June 22, 2016), patients who were still being followed at that date were administratively discontinued from the study. Therefore, the duration of participation in the LTE depended on the date of enrollment, ranging from 52 to 130 weeks (2.5 years), during which time patients received from 13 to 32 injections of AL. The AL dose remained fixed during both the 52-week and LTE safety studies; no AL dose adjustments were permitted.
Click figure to enlarge
Additional medication management. Anticholinergics and antihistamines were permitted for the treatment of extrapyramidal symptoms (EPS). β-blockers (eg, propranolol) were allowed for akathisia, and benzodiazepines could be given for akathisia, EPS, insomnia, agitation, or anxiety. Although ongoing concomitant treatment with other antipsychotics was not allowed after the first 4 weeks of the 52-week safety study, it was used occasionally thereafter as a rescue treatment. Because oral antipsychotics were allowed as part of the initial cross-taper, when patients were switching from an oral antipsychotic to AL, data from the first 4 weeks are excluded when presenting data on the concomitant antipsychotic medications.
Patients
Inclusion criteria for patients entering the 52-week study are described in detail in Nasrallah et al.6 Patients could enter the 52-week study after completing the 12-week phase 3 pivotal efficacy study (ALK9072-003; ClinicalTrials.gov identifier: NCT01469039; EudraCT Number: 2012-003445-15),5 continuing on their same blinded AL dose regimen, with those having been in the placebo group now being started on one of the AL doses based on their initial randomization status. Additional stable outpatients receiving oral antipsychotics were enrolled (n = 242) and were started on the higher-dose AL regimen to maximize the number of long-term safety patients receiving the highest exposure to AL. All patients continued on a fixed-dose AL regimen; no AL dosage strength adjustments were allowed at any time in either safety study.
Patients completing the 52-week study and requiring chronic treatment with an antipsychotic medication were eligible to enroll in the LTE unless they had a clinically relevant laboratory, vital sign, or electrocardiogram (ECG) abnormality; missed more than 1 scheduled visit; or required use of a prohibited concomitant medication during the 52-week study.
Safety Assessments
Safety in the combined 52-week and LTE studies over 180 weeks (3.5 years) was assessed by baseline and endpoint physical and laboratory examinations. Ongoing safety was assessed by standard AE reporting methods associated with accepted Good Clinical Practice standards. Other repeated, routine safety measures included assessment of injection site reactions, vital signs (including weight), suicide screening using the Columbia-Suicide Severity Rating Scale (C-SSRS),9 and movement disorder scales (Abnormal Involuntary Movement Scale [AIMS],10 Barnes-Akathisia Rating Scale [BARS],11 and Simpson-Angus Scale [SAS]12) for the duration of both studies.
- Two consecutive long-term extension studies assessed aripiprazole lauroxil safety and symptom improvement (441 mg or 882 mg intramuscularly every 4 weeks) for up to 3.5 years.
- Safety results were consistent with those of oral aripiprazole; no AE trends occurred with any regularity after 3 months treatment.
- Post hoc analysis (weeks 12-124) revealed clinically meaningful symptom improvement as measured by Positive and Negative Syndrome Scale total score reductions.
Data on all adverse events (AEs) were collected beginning when patients provided informed consent for entry into the 52-week study6 (eg, baseline at week 0) and continuing for up to 180 weeks (3.5 years) thereafter. For the current analysis, any report based on investigator judgment of a new AE from the first AL injection onward was included. Rates of AEs, serious AEs (SAEs), other safety outcomes, and all-cause hospitalization were summarized by AL dose group (441 mg vs 882 mg). AEs reported by ≥ 2% of patients were summarized. Reasons for study discontinuation were collected. Because many of the safety findings of the first 52 weeks have already been reported,6 in this article we report certain AEs by occurrence rates for the first 52 weeks and separately for the safety follow-up after 52 weeks until study completion.
Post Hoc Analyses
Post hoc analyses assessing durability of effect and symptom trajectory were based on a total follow-up duration of 112 weeks. The starting point for the post hoc time period was selected to minimize possible confounding effects of different AL exposure histories based on when patients were enrolled. Therefore, the post hoc period was started at the week 12 visit of the 52-week study. By this visit, all patients had received ≥ 3 AL injections and were considered stabilized on their AL regimen. The post hoc analysis endpoint was the week 124 visit of the combined 52-week and LTE studies and was chosen, based on a drop-off in sample size after this time (eg, week 124, n = 211; following visit, n = 94), because of the scheduled closure of the LTE study in June 2016. Therefore, the post hoc analysis period ranged from weeks 12 to 124 and covered more than 2 years of AL exposure (40 weeks during the 52-week study and 72 weeks during the LTE; Figure 1), during which time patients received up to 28 monthly AL injections.
Durability of effect was assessed over 112 weeks (ie, week 12 stabilization point to week 124) by time to medication-related discontinuation (due to AE or lack of efficacy) and all-cause discontinuation. Clinical symptom trajectory was assessed using the Positive and Negative Syndrome Scale (PANSS) total (PANSST) score13 and the PANSS Positive, Negative, and General Psychopathology subscale scores. Illness severity was assessed based on Clinical Global Impressions-Severity of Illness scale (CGI-S)14 scores. The PANSS and CGI-S were administered at weeks 12, 20, 32, 44, 48, 64, 76, 88, 100, 112, and 124.
Time to all-cause and medication-related discontinuations was estimated using the Kaplan-Meier method. Patients who completed the 52-week study but did not enter the LTE were censored at the 52-week time point. Changes in PANSST and subscale scores and CGI-S scores from the week 12 assessment visit to the patient’s last assessment visit were analyzed using a mixed-effects model for repeated measures (MMRM) by dose group. The model included the dose group (starting at the week 12 assessment), visit, and interaction terms of visit and dose group. The autoregressive 1 covariance structure was used to model the between-visits variance. Subgroup analyses in PANSST score changes, examining the effects of sex and age, were performed using similar models including sex (or age), visits, and an interaction term for sex (or age) and visits.
RESULTS
Baseline Characteristics and Patient Disposition in the Combined 52-Week and LTE Studies
The majority of patients in the safety population (n = 478) for the two studies were men (57.5%) and white (63.8%) and had a mean age of 39.4 years (range, 18-69 years). At baseline, the aggregate mean (SD) weight of patients participating in the two studies was 78.1 (18.1) kg, with a mean (SD) body mass index of 27.0 (5.3) kg/m2. Of the 478 patients entering the 52-week study, 326 completed the treatment period; 291 subsequently entered the LTE, and 259 completed it. Patient disposition through the 52-week safety study and the LTE study in aggregate, along with reasons for study discontinuation, are presented in Supplementary Table 1.
Adverse Events
AEs were reported in 275 (57.5%) of 478 patients across the two studies, including 52.7% of patients in the 441-mg dose group and 59.0% in the 882-mg dose group (Supplementary Table 1). The majority of AEs (89.8%) were mild or moderate in severity, and most AEs (n = 241) occurred during the initial 52-week safety study. The most common AEs across both studies were insomnia (12.8% [441 mg, 7.3%; 882 mg, 14.4%]), headache (6.7% [441 mg, 10.9%; 882 mg, 5.4%]), increased weight (6.1% [441 mg, 9.1%; 882 mg, 5.2%]), anxiety (5.6% [441 mg, 3.6%; 882 mg, 6.3%]), and nasopharyngitis (5.2% [441 mg, 7.3%; 882 mg, 4.6%]) (Supplementary Table 1). Overall, 34 patients discontinued because of AEs across the studies, including 28 during the 52-week study6 and 6 during the LTE. Exacerbation/worsening of schizophrenia (n = 14 [2.9%]), exacerbation of psychotic disorder (n = 2 [0.4%]), and akathisia (n = 2 [0.4%]) were the most common AEs leading to discontinuation.
SAEs occurred in 18 patients (3.8%) in aggregate across the two studies. There were 15 patients with a SAE occurring during the initial 52-week safety study described in Nasrallah et al.6 After 52 weeks, 4 patients experienced a total of 7 SAEs (1 patient had SAEs of lung mass and lung cancer; 1 had pneumonia and sepsis; 1 had suicidal ideation and, at a later date, pancreatitis; and 1 had exacerbation/worsening of schizophrenia). Two patients discontinued the study owing to SAEs (lung cancer and exacerbation/worsening of schizophrenia). None of the SAEs were considered by the investigator to be treatment-related, and none resulted in death.
Other Safety Topics
Injection site reactions. Over the course of these two consecutive safety studies, 24 (5.0%) of 478 patients had ≥ 1 injection site reaction (ISR) AE (441 mg, 1 [0.9%]; 882 mg, 23 [6.3%]). Of these, 23 (95.8%) were reported during the 52-week safety study6; most were mild in severity. Of 478 patients, 4 (0.8%) had moderate injection site pain and 1 (0.2%) had severe injection site pain. In the LTE, 3 (1.0%) of 291 enrolled patients experienced an ISR of injection site pain (mild severity in 2 and moderate severity in 1) after the first year.
Extrapyramidal symptoms including akathisia. EPS were assessed in two ways: by AE reporting and by assessment of standard movement disorder scales. AEs associated with EPS occurred in 61 patients (12.8% [441 mg, 9.1%; 882 mg, 13.9%]); all were mild or moderate in severity, and 45 (73.8%) of these were previously reported during the 52-week study.6 After the first year of treatment, the most commonly reported EPS-associated AEs were akathisia in 2 patients (1 each in the 441-mg and 882-mg groups) and tremor in 8 patients (1 in the 441-mg group and 7 in the 882-mg group).
Weight change. Patients gained a mean of 0.7 kg from their 12-month assessment to their last assessment. As already reported,6 a first year categorical weight increase of ≥ 7% occurred in 88 (18.4%) of 478 patients and a ≥ 7% weight decrease occurred in 59 (12.3%). After the first year, 26 (8.9%) of 291 patients gained ≥ 7% from their 12-month weight and 15 (5.2%) lost ≥ 7% from their 12-month weight. Combining both safety studies into a single follow-up, treatment with AL was associated with a mean (SD) body weight increase from baseline of 1.2 (6.6) kg overall in those continuing treatment.
Prolactin. At baseline, the mean (SD) plasma prolactin concentration was 20.6 (30.3) ng/mL (13.3 [15.8] ng/mL in men and 30.4 [40.6] ng/mL in women). Over the course of both studies, the mean (SD) prolactin concentration change from baseline to the last assessment was −11.1 (32.9) ng/mL overall (−6.7 [16.5] ng/mL in men and −16.5 [45.2] ng/mL in women).
In total, 7 patients had AEs of increased blood prolactin concentration (3 in the 441-mg group and 4 in the 882-mg group) and 8 had an AE of hyperprolactinemia (4 each in the 441-mg and 881-mg groups) over the course of the two studies; no patient discontinued owing to these AEs. One additional patient had an AE of hyperprolactinemia during the follow-up period. Overall, 8 patients (1.7%) reported prolactin-related AEs (decreased libido [n = 3], dysmenorrhea [n = 3], amenorrhea [n = 1], and menstrual disorder [n = 1]); 5 of these occurred in the 52-week safety study.6 There were no reports of gynecomastia, galactorrhea, breast tenderness, or erectile dysfunction. Nine patients, including 2 (1.3%) of 151 men and 7 (6.4%) of 110 women with prolactin concentration within normal limits at baseline, had prolactin elevations that were ≥ 3 times the upper limit of normal at any postbaseline assessment across the two studies, representing an additional 7 patients (2 men and 5 women) over that observed in the 52-week study.6
There were no clinically meaningful findings in physical examinations, vital signs, laboratory values (including those assessing metabolic parameters, liver and kidney function, or hematology indices), ECGs, or suicidal ideation (C-SSRS) results over the course of the two studies.
Other Psychiatric Medications
Three commonly prescribed adjunctive or concomitant therapies provided during the first year of treatment included benzodiazepines (n = 96 [20.1%]), adjunctive anticholinergics for EPS or akathisia (n = 65 [13.6%]), and oral antipsychotics (n = 15 [3.2%]), excluding the initial crossover period. For patients entering into > 1 year of AL treatment, 9 (3.1%) of 286 received ≥ 1 dose of antipsychotic medication at some point between week 52 and their last study visit.
Post Hoc Analyses
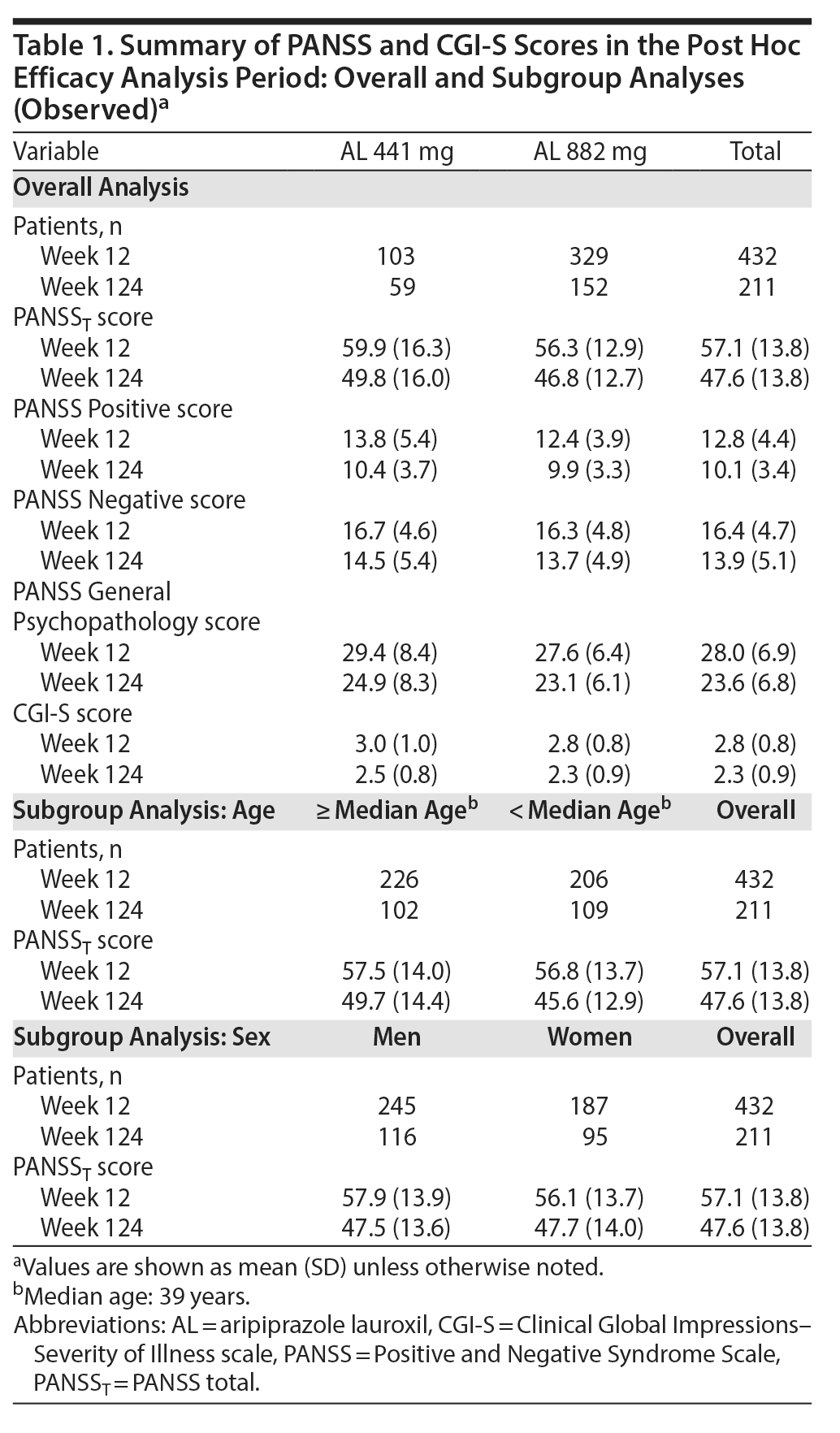
Of the 478 patients who entered the 52-week study, 432 (441 mg, n = 103; 882 mg, n = 329) completed the 12-week assessments and were included in the post hoc analyses of durability of effect, rehospitalization, trajectory of clinical symptoms, and trajectory of illness severity. Of these, 211 completed the post hoc follow-up period.
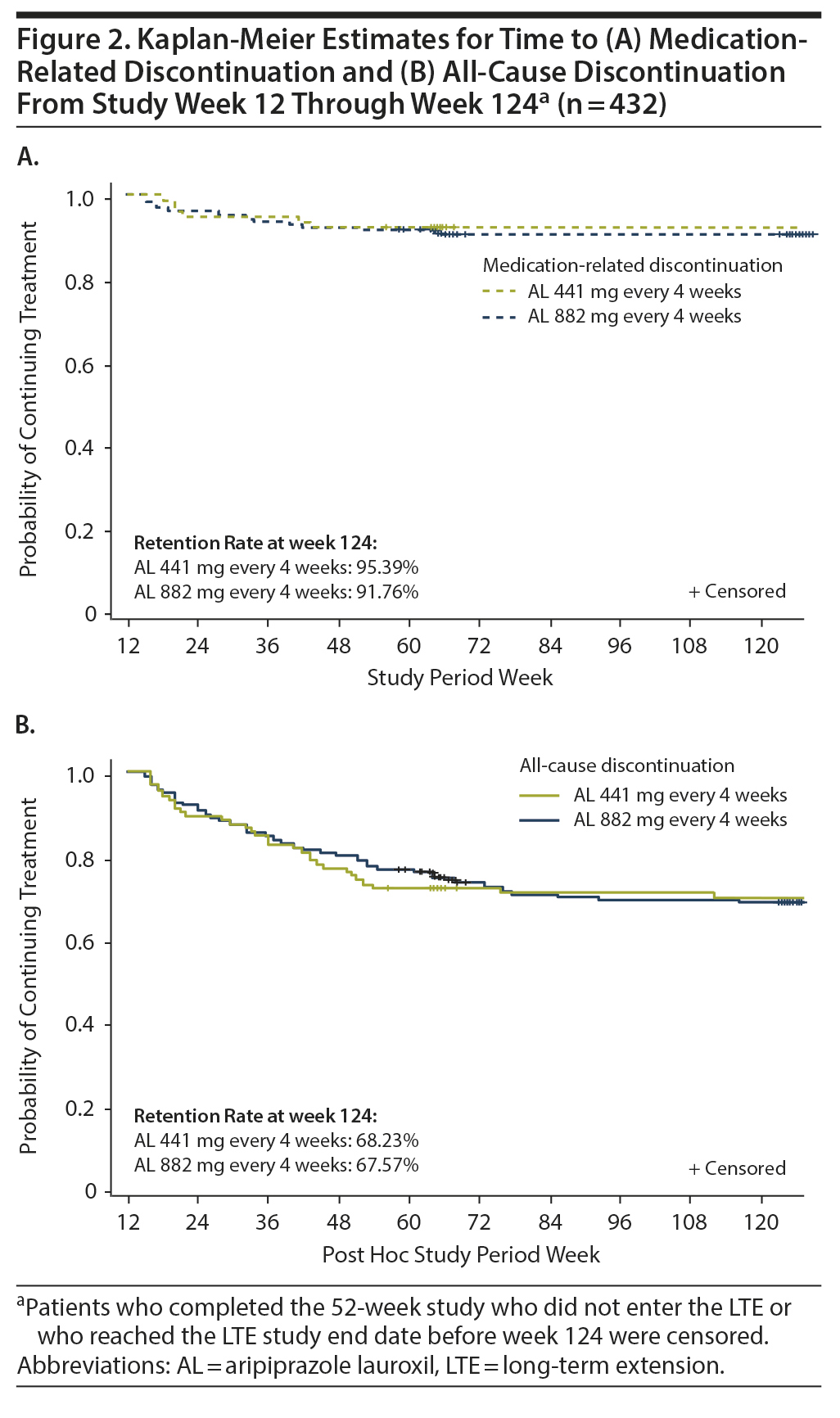
Durability of treatment effect. Kaplan-Meier estimates for medication-related discontinuation (Figure 2A) and for all-cause discontinuation (Figure 2B) were similar for the AL 441-mg and 882-mg dose groups. Over the 12- to 124-week post hoc analysis period, discontinuations due to medication effects occurred in ~5% (AL 441 mg) to ~8% (AL 882 mg) of patients, while all-cause discontinuations occurred in ~32% in each treatment group.
Click figure to enlarge
Rehospitalization. The overall all-cause hospitalization rate from week 12 to the end of the LTE study was 5.6% (24/432), with 3.9% of patients (4/103) from the 441-mg group and 6.1% (20/329) from the 882-mg group being hospitalized. Exacerbation of schizophrenia (n = 8 [1.9%]) and chronic obstructive pulmonary disease (n = 2 [0.5%]) were the only reasons for rehospitalization that occurred in > 1 patient in any treatment group.
Trajectory of clinical symptoms. In the 52-week safety study previously reported, PANSST scores improved from 63.6 at the week 12 visit to 52.0 (441 mg) and 63.4 to 52.0 (882 mg), representing LS mean changes in PANSST scores of −8.1 and −7.2 points, respectively.15
A post hoc analysis assessing durability of effect in patients who continued into the 52-week safety study revealed further schizophrenia symptom improvement with continued AL treatment. In this post hoc analysis, despite having fairly low least-squares (LS) mean PANSST scores at week 12 baseline (AL 441 mg, 59.9; AL 882 mg, 56.3; Table 1), further significant reductions in LS mean PANSST scores of −5.5 and −5.0, respectively, were observed between weeks 12 and 124, indicating continued symptom improvement beyond the initial year of treatment (Figure 3A). The LS mean (95% CI) difference between treatment groups was 2.8 (−0.9 to 6.4), indicating no significant dose-dependent effect. A similar pattern of improvement was observed on the PANSS Positive and Negative subscales (Figure 3B and Figure 3C, respectively) and in General Psychopathology subscale scores (Supplementary Figure 1). In subgroup analyses, no significant difference was observed in the magnitude of improvement in PANSST over the post hoc period for patients older versus younger than the median age (> 39 vs ≤ 39 years) or for men versus women.
Click figure to enlarge
Trajectory of illness severity. Clinically relevant improvement in CGI-S total scores from weeks 12 to 124 was also observed in both AL dose groups. Mean (SD) week 12 CGI-S scores were 3.0 (0.97) and 2.8 (0.79) in the 441-mg and 882-mg groups, respectively (Table 1). At week 124, the LS mean (SE) CGI-S scores were 2.6 (0.10) in the 441-mg group and 2.5 (0.06) in the 882-mg group. The LS mean difference and associated 95% CIs for the change in CGI-S between the AL 441-mg and 882-mg treatment groups from weeks 12 to 124 was 0.1 (−0.1 to 0.4), indicating no between-group difference (Figure 3D).
Click figure to enlarge
DISCUSSION
Results for patients who received AL 441-mg or 882-mg injections every 4 weeks for up to 180 weeks (3.5 years) further inform the safety and tolerability of long-term treatment with AL in patients with schizophrenia. Continued therapeutic efficacy was observed from the 12-week stabilization point through week 124 in the post hoc follow-up period. In this analysis, the estimated probability of medication-related discontinuation was low.
The AE profile for AL in the aggregate study analysis lasting up to 180 weeks (3.5 years) was similar to that reported in the 52-week safety study.6 The most common AEs reported in the aggregate analysis, including insomnia, headache, increased weight, and anxiety, were also among those reported during the initial 52-week safety study.6 The overall rate of AEs (57.5%) was comparable to that found in the 52-week study alone (50.4%), and rates of SAEs were also comparable (3.8% vs 3.1%, respectively).6 The incidence of increased weight as an AE in the current analysis (6.1%) was similar to that reported in the 52-week study (5.0%).6 The proportion of patients with weight gains of ≥ 7% increased with longer-term exposure to AL (18.4% in the 52-week study and 23.8% in this combined analysis with exposure of up to 180 weeks [3.5 years]).
The incidence of ISRs was low in the current aggregate analysis (5%) and comparable to that found in the pivotal phase 3 AL study.5 Of note, 23 of the 24 patients reporting ISRs did so during the initial 52-week safety study.6
In the aggregate safety analysis combining both studies, less than 5% of patients overall reported akathisia as an AE (441 mg, 5/110 [4.5%]; 882 mg, 18/368 [4.9%]), representing an additional 3.6% in the AL 441-mg group and an additional 0.3% in the 882-mg group over that reported in the 52-week study (441 mg, 0.9%; 882 mg, 4.6%)6 and lower proportions than were observed in the 12-week pivotal study (441 mg, 11.6%; 882 mg, 11.5%).5 Most episodes of akathisia in the 52-week study occurred early, when AL treatment was being initiated in patients who had been assigned to placebo in the 12-week efficacy study or in those who were enrolled de novo (all of whom were assigned to AL 882 mg). Similarly, over 75% of akathisia events in the 12-week efficacy study occurred before the second AL injection; most occurred in the first 3 weeks during supplementation with oral aripiprazole.5
Acute treatment with AL 441 or 882 mg is associated with significant improvement in schizophrenia symptoms,5 and this 2-year follow-up post hoc analysis demonstrated continued improvement in symptoms and severity of illness during long-term AL treatment. In the 12-week efficacy study, change from baseline in placebo-adjusted PANSST scores was −10.9 and −11.9 for the 441-mg and 882-mg dose groups, respectively. A post hoc analysis assessing durability of response in patients who continued into the 52-week safety study revealed further schizophrenia symptom improvement with continued AL treatment. The PANSST scores improved from 63.6 to 52.0 (441 mg) and 63.4 to 52.0 (882 mg) during the 52-week study, representing LS mean changes in PANSST scores of −8.1 and −7.2 points, respectively (both P < .0001).15 In the current analysis, significant reductions in PANSST score of −5.5 and −5.0 between weeks 12 and 124 were observed for the 441-mg and 882-mg dose groups, respectively, indicating continued symptom improvement beyond the initial year of treatment (Figure 3A). No difference between the dose groups in efficacy was apparent as measured by changes in PANSS or CGI-S scores, which is not unexpected as no additional benefit for higher therapeutic doses has been observed with oral aripiprazole16 or other antipsychotic medications.17
The current analysis has several limitations. First, the inclusion of patients who successfully completed the 12-week pivotal efficacy and safety study may limit the generalizability of the results to a broader population of patients with schizophrenia who would not meet the study entry criteria. In both the 52-week study and LTE, the efficacy of AL was evaluated at fixed assigned doses with no dose adjustments, contrary to what commonly occurs in clinical practice. In addition, the assignment of all patients enrolled de novo into the 882-mg group confounded interpretation of any differences in tolerability, durability of effect, or symptom trajectory between AL dose groups. All patients in the 441-mg group had previously received 12 weeks of exposure to AL in the pivotal study, and prior exposure itself may be associated with lower rates of AEs that frequently occur at treatment initiation, such as ISR or akathisia. Also, although the week 12 stabilization point was selected to minimize the effects of the differences in AL exposure between enrolled patients with different treatment histories, prior treatment (double-blind AL or placebo or stable oral antipsychotic medication) may have affected longer-term outcomes in the post hoc analysis. Additionally, starting the post hoc analysis at week 12 of the 52-week safety study may have biased the analysis toward a more stable cohort of patients. Furthermore, the attrition of patients who had poor tolerability or lack of clinical efficacy can result in survivor biases, with the remaining patients having relatively better symptom response or fewer AEs than those who discontinued earlier. With that limitation in mind, the lack of any new or unexpected AEs in this patient population may be reassuring to clinicians concerned about possible delayed safety issues due to the nature of persistence of plasma concentrations associated with long-acting therapies. Regarding the trajectory of gradual but significant time trends in symptom improvement based on PANSS scores over time, MMRM analysis may reduce the survivor bias effect because it takes into account the course of symptoms in all patients, including those who discontinued before their last assessment. The table reporting all AEs for all patients partly addresses the latter concern. Although we acknowledge that the data reported here overlap with previously published data from the 52-week safety study, our primary goal was to assess the pattern of safety and durability of effect well beyond the 1-year period previously reported.6
CONCLUSIONS
In this aggregate safety analysis detailing up to 180 weeks (3.5 years) of safety and tolerability data from sequential long-term trials, AL 441 mg and 882 mg every 4 weeks was generally well tolerated. The AL safety profile observed over a period of up to 180 weeks (3.5 years) of treatment was consistent with that of oral aripiprazole and with the previously reported 52-week AL safety study results. No unexpected safety concerns were identified during the 180-week (3.5-year) follow-up period. Continuous efficacy data examined post hoc demonstrated that schizophrenia symptoms and severity of illness improved over the course of the 112-week study period, with no apparent dose effect. Results of this analysis demonstrate the tolerability and continued therapeutic efficacy of long-term treatment with AL 441 mg and 882 mg given every 4 weeks in adult patients with schizophrenia.
Submitted: March 15, 2019; accepted June 17, 2020.
Published online: August 18, 2020.
Potential conflicts of interest: Dr Lauriello discloses the following: advisory board member for Otsuka, Indivior, Osmotica, and Alkermes, Inc; speaker at investigator’s meeting for Roche; Continuing Medical Education (CME) activity for CME Vindico Medical Education/Psych Forum Webinar and CME Institute Webinar; travel funds from Alkermes, Inc; and event monitoring board member for Alkermes, Inc. Drs Claxton and Du are employees of Alkermes, Inc, and may own stock/options in the company. Dr Weiden is a former employee of Alkermes, Inc, and may own stock/options in the company.
Funding/support: This study was sponsored by Alkermes, Inc.
Role of the sponsor: The study sponsor was involved in the design, collection, and analysis of the data. Interpretation of the results was by the authors, and the decision to submit the manuscript for publication was made by the authors.
Previous presentation: American Society of Clinical Psychopharmacology Annual Meeting; May 28-June 1, 2018; Miami, Florida ▪ 31st Annual Psych Congress; October 25-28, 2018; Orlando, Florida ▪ Neuroscience Education Institute Congress; November 7-11, 2018; Orlando, Florida.
Acknowledgments: Medical writing and editorial support were provided by Kathleen Dorries, PhD, and John H. Simmons, MD, of Peloton Advantage, LLC (Parsippany, New Jersey), an OPEN Health company, and funded by Alkermes, Inc.
Supplementary material: See accompanying pages.
REFERENCES
1.Hasan A, Falkai P, Wobrock T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44. PubMed CrossRef
2.Weiden PJ, Kozma C, Grogg A, et al. Partial compliance and risk of rehospitalization among California Medicaid patients with schizophrenia. Psychiatr Serv. 2004;55(8):886-891. PubMed CrossRef
3.Kane JM. Review of treatments that can ameliorate nonadherence in patients with schizophrenia. J Clin Psychiatry. 2006;67(suppl 5):9-14. PubMed
4.Aristada [package insert]. Waltham, MA: Alkermes, Inc; 2018.
5.Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090. PubMed CrossRef
6.Nasrallah HA, Aquila R, Du Y, et al. Long-term safety and tolerability of aripiprazole lauroxil in patients with schizophrenia. CNS Spectr. 2019;24(4):395-403. PubMed CrossRef
7.WMA Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects. World Medical Association website. https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/. 2013. Accessed September 24, 2018.
8.Guideline for good clinical practice E6(R2). European Medicines Agency website. https://www.fda.gov/media/93884/download. 2012. Accessed September 24, 2018. >
9.Posner K, Brent D, Lucas C, et al. Columbia-Suicide Severity Rating Scale (C-SSRS) since last visit. Columbia University website. https://cssrs.columbia.edu/wp-content/uploads/C-SSRS-1-14-09-SinceLastVisit_AU5.1_eng-USori-1.pdf. 2009. Accessed September 24, 2018.
10.Guy W. Abnormal Involuntary Movement Scale (AIMS). ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: National Institute of Mental Health, National Institutes of Health; 1976:534-537.
11.Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672-676. PubMed CrossRef
12.Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl. 1970;212:11-19. PubMed CrossRef
13.Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261-276. PubMed CrossRef
14.Guy W. ECDEU Assessment Manual for Psychopharmacology. Revised Edition. Washington, DC: US Department of Health, Education, and Welfare; 1976.
15.McEvoy JP, Risinger R, Mykhnyak S, et al. Durability of therapeutic response with long-term aripiprazole lauroxil treatment following successful resolution of an acute episode of schizophrenia. J Clin Psychiatry. 2017;78(8):1103-1109. PubMed CrossRef
16.Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771. PubMed CrossRef
17.Samara MT, Klupp E, Helfer B, et al. Increasing antipsychotic dose for non response in schizophrenia. Cochrane Database Syst Rev. 2018;5:CD011883. PubMed
This PDF is free for all visitors!





