A 10-Month, Open-Label Evaluation of Desvenlafaxine in Outpatients With Major Depressive Disorder
Background: The primary objective was to evaluate the long-term safety of desvenlafaxine (administered as desvenlafaxine succinate) during open-label treatment in adult outpatients with a primary DSM-IV diagnosis of major depressive disorder (MDD).
Method: Depressed adult outpatients (≥ 18 years) who had completed 8-week, double-blind therapy (desvenlafaxine, venlafaxine extended release, or placebo) in a phase 3 study of desvenlafaxine for MDD received up to 10 months of open-label treatment with flexible-dose desvenlafaxine (200 to 400 mg/d). Safety assessments included physical examination, measurement of weight and vital signs, laboratory determinations, and 12-lead electrocardiogram recordings. Adverse events (AEs) and discontinuations due to AEs were monitored throughout the trial. The primary efficacy outcome was mean change from baseline on 17-item Hamilton Depression Rating Scale (HDRS-17) total score. The trial was conducted from August 2003 to March 2006.
Results: The safety population included 1,395 patients who took at least 1 dose of open-label desvenlafaxine. Treatment-emergent AEs were reported by 1,238 of 1,395 patients (89%) during the open-label, on-therapy period. Treatment-emergent AEs reported by 10% or more patients were headache, nausea, hyperhidrosis, dizziness, dry mouth, insomnia, upper respiratory infection, nasopharyngitis, and fatigue. Adverse events were the primary reason for study discontinuation in 296 of 1,395 patients (21%). Ten patients (< 1%) had serious AEs that were considered possibly, probably, or definitely related to the study drug during the on-therapy period. No deaths occurred during the study.
Conclusions: Desvenlafaxine can be safely administered for up to 12 months. No new safety findings were observed in this study.
Trial Registration: clinicaltrials.gov Identifier: NCT01309542
Prim Care Companion CNS Disord 2011;13(2):e1-e10
© Copyright 2011 Physicians Postgraduate Press, Inc.
Submitted: March 5, 2010; accepted June 10, 2010
Published online: April 7, 2011 (doi:10.4088/PCC.10m00977).
Corresponding author: Karen A. Tourian, MD, CR&D Neuroscience, Wyeth Pharmaceuticals France, CÅ“ur Défense-Tour A-La Défense 4, 110, Esplanade du Générale de Gaulle, 92931 Paris, La Défense Cedex, France ([email protected]).
Notice of correction 6/8/2011: Fasting triglyceride levels in Table 3 have been corrected. Baseline mean = 134.46 mg/dL; mean change from baseline = 10.48 mg/dL.
Major depressive disorder (MDD) is a seriously impairing illness associated with substantial disability.1-5 Depression follows a chronic course, with sustained remission in approximately 20% to 35% of cases.6-8 Approximately 50% of patients with MDD experience a second episode of depression within 5 to 10 years of recovery.8 To reduce the risk of recurrence of depressive episodes, antidepressant treatment is recommended for at least 16 to 20 weeks after acute-phase treatment is completed and for up to 2 years in patients at risk for recurrence of depressive episodes.9-11 Thus, there remains a need for antidepressant agents that are effective, safe, and well tolerated for both acute and long-term management of MDD.
Clinical Points
- Because antidepressant therapy is recommended to continue for a minimum of 16-20 weeks after acute-phase treatment, understanding the long-term safety and tolerability of approved treatments is important.
- Desvenlafaxine treatment, even at the higher end of the dose range, was safely administered for up to 12 months.
- The recommended therapeutic dose of desvenlafaxine remains 50 mg/d, and clinicians should consider long-term treatment as warranted for the individual patient.
The serotonin-norepinephrine reuptake inhibitor (SNRI) desvenlafaxine (administered as desvenlafaxine succinate) is the major active metabolite of the antidepressant venlafaxine.12,13 The metabolic profile of desvenlafaxine suggests a low risk of drug-drug interactions. Desvenlafaxine has minimal inhibitory effects on cytochrome P450 (CYP) 2D6 and no clinically relevant effect on CYP2D6 metabolism,14-16 low protein binding (30%),13 and no interaction with P-glycoprotein.16
Short-term safety, tolerability, and efficacy for the treatment of MDD have been demonstrated for desvenlafaxine 50-, 100-, 200-, and 400-mg/d doses in phase 3, double-blind, randomized, placebo-controlled, fixed-dose, 8-week MDD trials.17-20 The recommended therapeutic dose of desvenlafaxine is 50 mg/d, administered as a single 50-mg tablet once daily.13 Integrated efficacy and safety analyses using the complete set of 5 fixed-dose registration trials of desvenlafaxine for MDD reported that the 50-mg/d dose is associated with the fewest discontinuations due to adverse events (AEs) at a rate comparable to placebo (approximately 4% for both) and that there is no evidence that doses greater than 50 mg/d confer any additional benefit.21,22
Desvenlafaxine treatment of MDD has predominantly been studied in short-term use, and few data are available to assess the long-term safety and tolerability of desvenlafaxine in the maintenance treatment of MDD. The primary objective of the current study was to evaluate the long-term safety of desvenlafaxine during open-label treatment in adult outpatients with a primary diagnosis of MDD. At the time this study was initiated, the 50-mg/d dose had not been investigated in short-term studies. This study used doses of desvenlafaxine that are at the higher end of the dose range, and thus any safety or tolerability issues are likely to be magnified. The secondary objective was to evaluate the long-term efficacy of desvenlafaxine for MDD.
METHOD
This phase 3, multicenter, open-label, flexible-dose, extension safety study enrolled adult outpatients with a primary diagnosis of MDD who had completed 1 of 6 randomized, double-blind, 8-week, phase 3, desvenlafaxine short-term MDD studies (study 304,23 306,17 308,18 309 and 317,24 or 32025). The trial was conducted from August 2003 to March 2006 in accordance with the US Food and Drug Administration (FDA) Code of Federal Regulations (21CFR, part 50),26 the International Conference on Harmonisation,27 and ethical principles based in the Declaration of Helsinki.28 The study was consistent with Good Clinical Practice29 and applicable regulatory requirements. All participants provided written, informed consent before enrollment.
Patients
Patients enrolled in the 10-month extension study were all adult outpatients (≥ 18 years of age) meeting Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition30 criteria for MDD who had completed 8-week, double-blind therapy in a phase 3 study of desvenlafaxine for MDD. Enrolled patients had no major protocol violations or study events during the lead-in, short-term studies17,18,23-25 that precluded entry into the long-term, open-label study. A response to short-term treatment was not an eligibility requirement for enrollment. Patients were excluded if they had clinically important abnormalities at the extension study baseline physical examination or any unresolved clinically important abnormalities recorded before day 56 in a double-blind, short-term, lead-in study. Patients also were excluded if they had clinically important medical disease or were judged to be at significant risk of suicide.
Study Design
All patients received open-label, flexible-dose desvenlafaxine (200 to 400 mg/d) for up to 10 months to allow up to 12 months of exposure to desvenlafaxine. In the double-blind, short-term trials contributing to the current study, patients received fixed- or flexible-dose desvenlafaxine 100 to 400 mg/d, venlafaxine extended release (ER) 75 to 225 mg/d, or placebo for 8 weeks. Prior treatment assignment was blinded at the time of entry into this study. Patients receiving desvenlafaxine or venlafaxine ER in the short-term trials were not tapered from the short-term medication before starting open-label desvenlafaxine. All patients, regardless of acute-phase treatment assignment, received desvenlafaxine 200 mg/d on days 1 through 7 of the current study, starting the day after their last on-therapy dose of short-term study drug. At week 1 and at each subsequent visit, the patients were evaluated and the investigator increased desvenlafaxine dose to 400 mg/d, if indicated, for efficacy. For patients who could not tolerate 400 mg/d, dosage was reduced to 200 mg/d.
Desvenlafaxine was tapered after the on-therapy period or at early withdrawal. Patients tapering from desvenlafaxine 400 mg/d received desvenlafaxine 200 mg/d for 7 days and then 100 mg/d for 7 days; patients tapering from desvenlafaxine 200 mg/d received 100 mg/d for 7 days. The taper period could be omitted or modified at the discretion of the investigator. Patients returned for a follow-up visit approximately 7 days either after their last tapered dose of desvenlafaxine or after their last on-therapy dose if the taper period was omitted.
Safety, Tolerability, and Efficacy Assessments
Baseline safety assessments, including physical examination, measurement of weight and vital signs, laboratory determinations, and 12-lead electrocardiogram (ECG) recordings, were obtained from the final on-therapy visit of the short-term studies. Changes from baseline in the short-term, lead-in studies also were examined. Spontaneously reported AEs, discontinuations due to AEs, and concomitant medications were monitored throughout the trial and during the poststudy taper period (the Discontinuation-Emergent Signs and Symptoms31 checklist was not used in the study because of lack of placebo control). Treatment-emergent and taper/poststudy-emergent AEs were classified based on the Medical Dictionary for Regulatory Activities32 and tabulated. Patient-level data, including serious adverse events (SAEs; categorized by standard regulatory definitions), noteworthy AEs (non-SAEs for which the sponsor required expedited reporting), and AEs of clinical importance, are reported using the Coding Symbols for the Thesaurus of Adverse Reaction Terms33 recorded in the study database. Weight and vital signs (supine and standing blood pressure, supine pulse rate) were measured at each study visit. Laboratory determinations (blood chemistry, hematology, and urinalysis) and ECG recordings were made at months 3, 6, and 10. A physical examination was conducted at month 10. For patients who withdrew early, all evaluations scheduled for month 10 were obtained on the last day on which the patient took a full dose of study medication or as soon as possible thereafter.
The primary efficacy outcome was mean change from baseline on the 17-item Hamilton Depression Rating Scale (HDRS-17)34 total score. Baseline HDRS-17 scores were obtained from the final on-therapy visit (acute-phase study day 56) of the respective short-term studies in which patients had participated before enrolling in this study. Change from acute-phase baseline (baseline visit for the short-term studies) was assessed in additional analyses. HDRS-17 total score was assessed at each on-therapy visit (weeks 1 and 2; months 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10); the HDRS-17 was not administered during tapering or at follow-up. The primary end point was the final evaluation; the secondary efficacy outcome was HDRS-17 remission rate (HDRS-17 total score ≤ 7) at final evaluation.
Statistical Analysis
The primary objective of the study was to assess the safety and tolerability of long-term desvenlafaxine treatment; therefore, a sample size estimate of approximately 1,500 patients was based on the number of patients available from short-term studies for obtaining adequate long-term safety data. Safety analyses were based on the safety population, which included all patients who took at least 1 dose of open-label study medication. The primary efficacy analysis was based on the intent-to-treat (ITT) population (all patients who had a baseline primary efficacy evaluation, took ≥ 1 dose of open-label study medication, and had ≥ 1 primary efficacy evaluation after the first dose of open-label study medication).
Changes in vital signs, laboratory assessments, and ECG findings from acute-phase and extension study baselines were summarized and assessed with t tests using the ≥ .05 significance level without adjustment for multiple tests. Mean changes in HDRS-17 total score from acute-phase baseline and from extension study baseline were assessed using the last-observation-carried-forward (LOCF) approach for handling missing data. The percentage of patients achieving HDRS-17 remission at each time point was calculated.
RESULTS
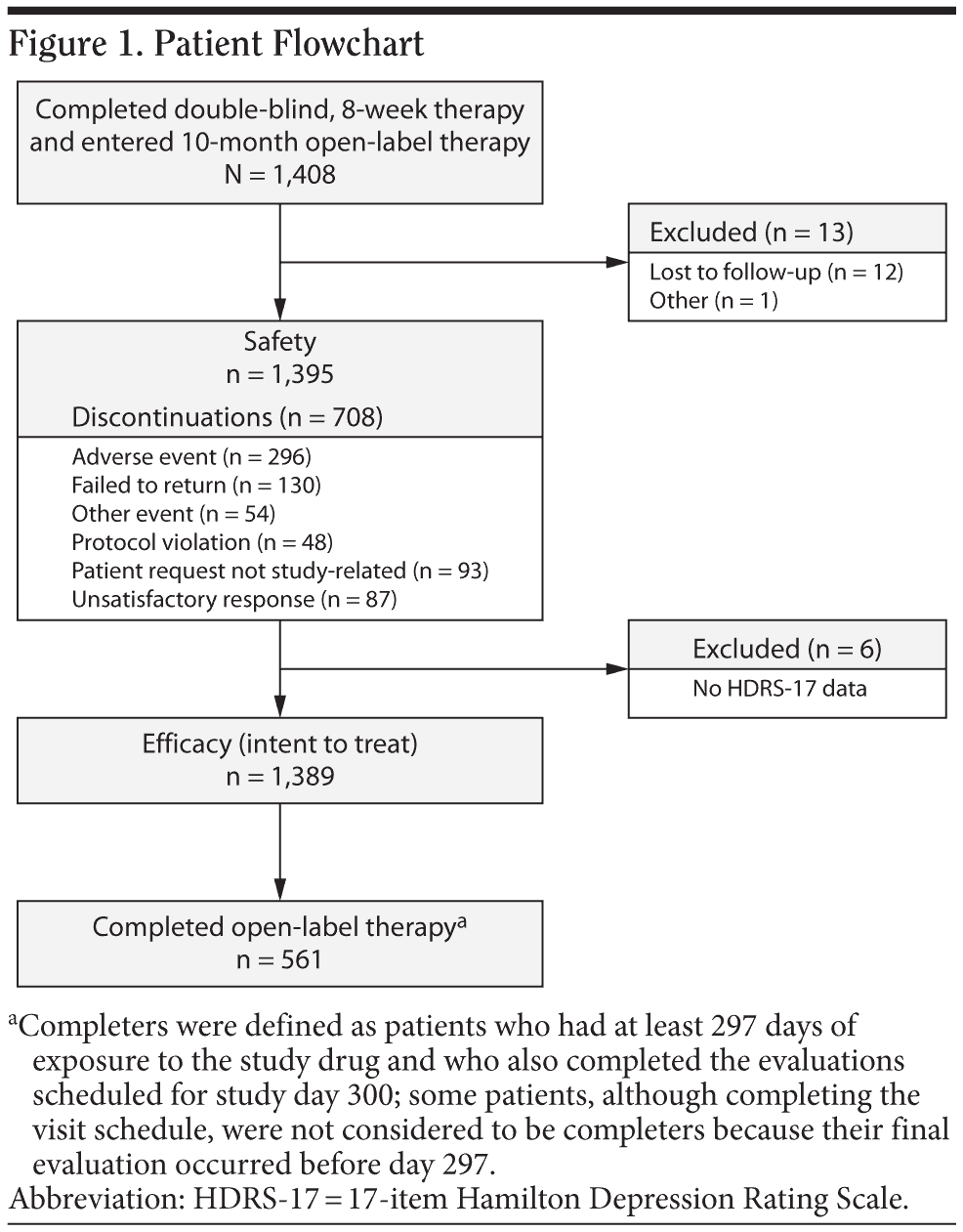
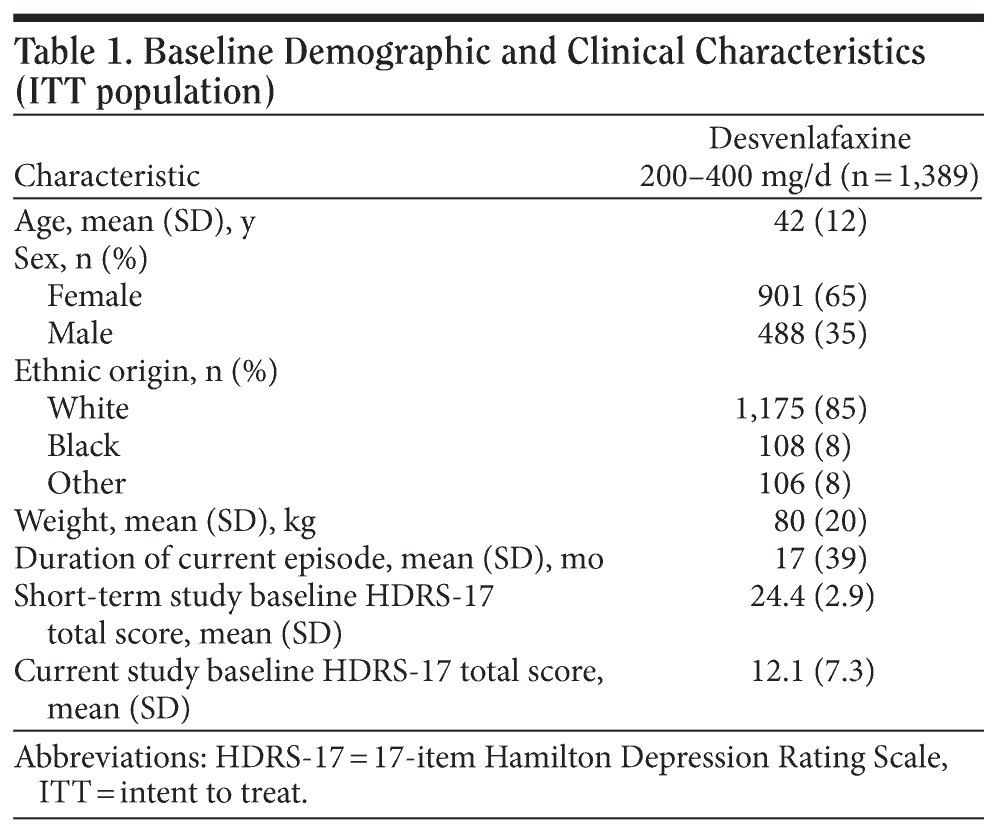
A total of 1,408 patients enrolled in the open-label extension study after completing 1 of the 6 short-term, double-blind trials (Figure 1). Of those, 13 patients were excluded from the safety population (n = 1,395): 12 were lost to follow-up after the baseline visit, and 1 returned all open-label study medication unused. Six patients in the safety population were excluded from the ITT efficacy population because they had no HDRS-17 evaluation after baseline; the ITT efficacy population included 1,389 patients. A total of 561 patients completed the study (had at least 297 days of open-label desvenlafaxine exposure and completed the month-10 evaluations). Additional patients completed the visit schedule, but were not considered completers because they had their final evaluation prior to day 297. Baseline patient demographic and clinical characteristics of the ITT population are shown in Table 1. During the on-therapy period (excluding the week-1 titration phase), the mean (± SD) daily desvenlafaxine dose for completers ranged from 246.9 mg/d (± 84.0) at week 2 to 298.4 (± 98.9) at month 7.
Click figure to enlarge
Click figure to enlarge
Safety and Tolerability
A total of 708 of 1,395 patients (51%) discontinued before completion of the study (Figure 1). Adverse events were the primary reason for study discontinuation in 296 of 1,395 patients (21%) during the on-therapy period. The AEs cited for early withdrawal by more than 1% of patients were nausea (49/1,395 [4%]), hypertension (36/1,395 [3%]), dizziness (35/1,395 [3%]), and insomnia (22/1,395 [2%]).
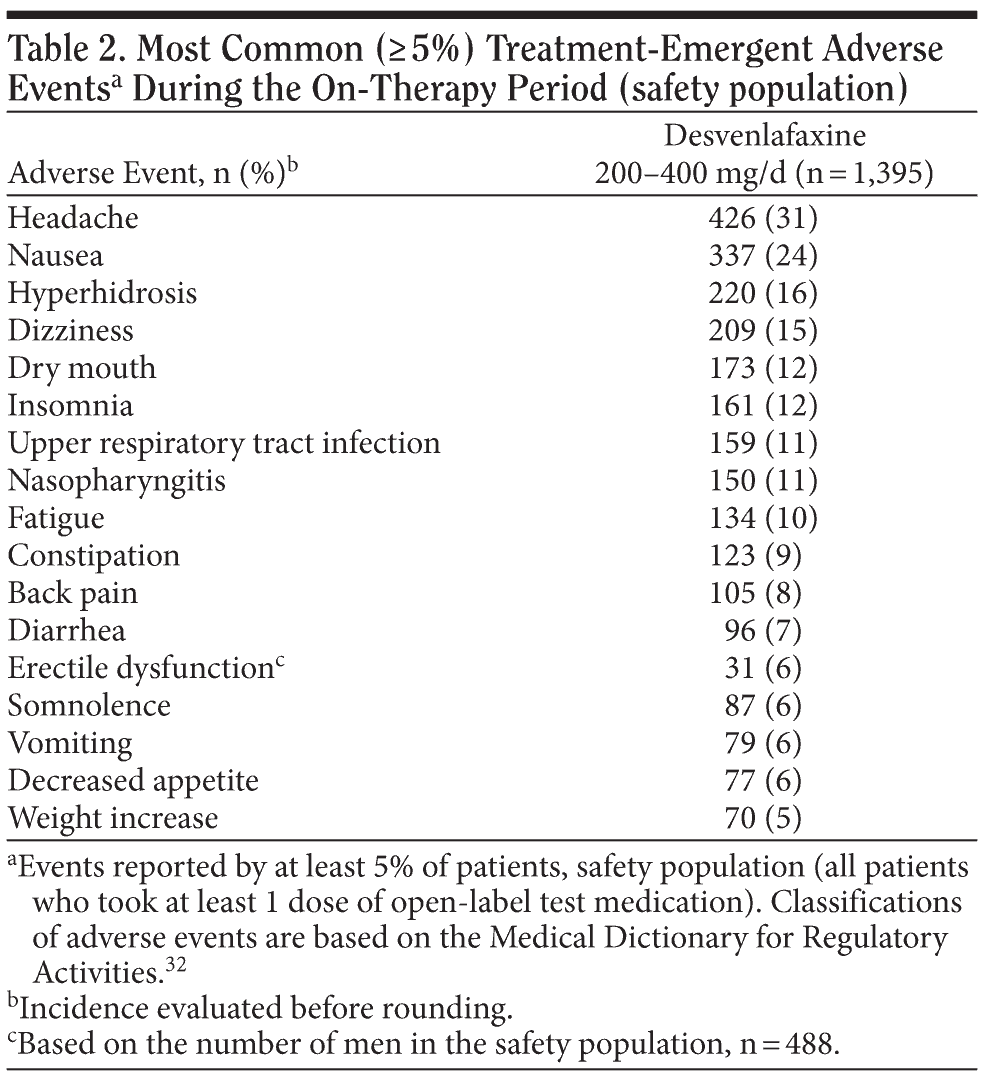
Adverse events. Treatment-emergent AEs were reported by 1,238 of the 1,395 patients (89%) during the open-label, on-therapy period. Most (78%) were mild or moderate in severity. The most common (incidence ≥ 5%) treatment-emergent AEs reported in this study are listed in Table 2. Treatment-emergent AEs reported by 10% or more patients were headache (31%), nausea (24%), hyperhidrosis (16%), dizziness (15%), dry mouth (12%), insomnia (12%), upper respiratory tract infection (11%), nasopharyngitis (11%), and fatigue (10%).
Click figure to enlarge
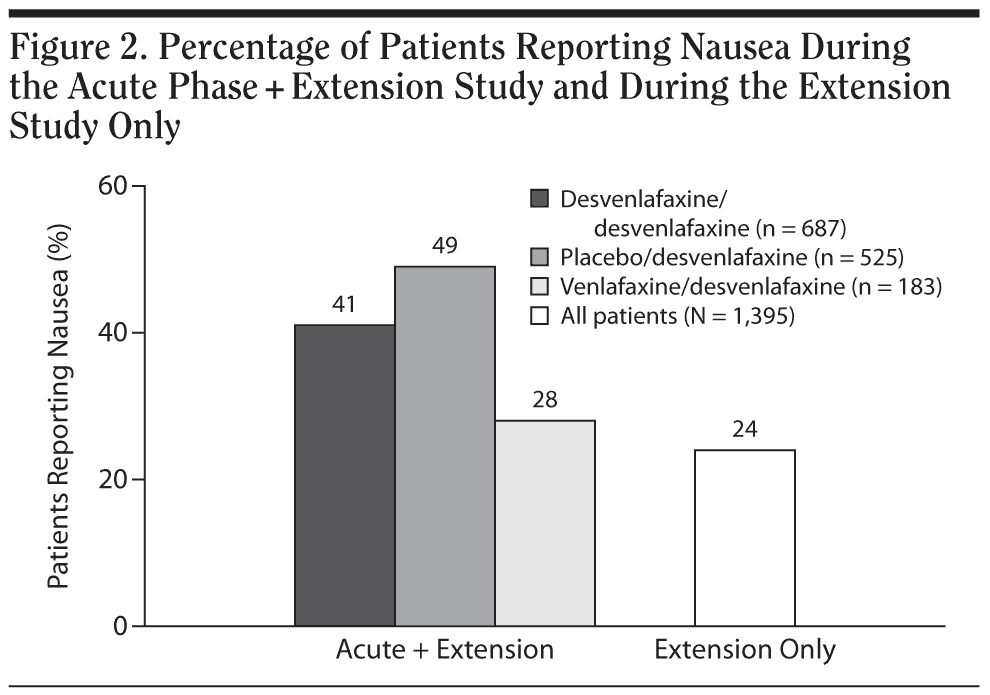
The percentage of patients who reported nausea from acute-phase baseline through the extension study was numerically greater for patients who had been previously assigned to double-blind, acute-phase placebo treatment (255/525 [49%]) compared with patients who received double-blind desvenlafaxine (281/687 [41%]) or venlafaxine ER (51/183 [28%]; Figure 2). Other common treatment-emergent AEs had similar rates among those groups. During the extension study, rates of spontaneously reported AEs related to sexual function were low, particularly for female patients: no AEs related to sexual function were reported by 5% or more of women during the extension study. Erectile dysfunction was reported by 31 of 488 male patients (6%); all other AEs related to sexual function were reported by less than 5% of men during the extension study.
Click figure to enlarge
A total of 1,249 of 1,395 patients (90%) were eligible to enter the taper period and to have the study medication tapered over a 2-week period after open-label treatment; 678 patients tapered per protocol, 146 patients had their taper period extended, 74 had their taper period shortened, and 351 had their taper period omitted at the discretion of the investigator. For the remaining 10% of patients, 144 withdrew early and 2 had no record of taper. Taper/poststudy-emergent AEs were reported by 584 of 1,395 patients (42%) in the safety population. The most common (incidence ≥ 5%) taper/poststudy-emergent AEs were dizziness (12%), nausea (9%), headache (8%), and withdrawal syndrome (5%). Consistent with how investigators were instructed to record syndromes consisting of multiple individual symptoms (eg, flu), the term withdrawal syndrome was used to describe related symptoms that occurred together as a group, in the judgment of the investigator, and may have included nausea, dizziness, and/or other symptoms.
Serious AEs and noteworthy AEs were reported by 65 of the 1,395 patients (< 5%) in the safety population. Noteworthy AEs reported during the on-therapy period included unintended pregnancies and 3 incidences of intentional overdose of study drug to increase efficacy (1 patient) or make up missed doses (2 patients). During the on-therapy period, 9 patients (< 1% of the safety population) had SAEs that were considered possibly, probably, or definitely related to the study drug. Those SAEs included 1 suicide attempt and 1 suicidal ideation (each considered possibly related to the study drug), asthenia, withdrawal syndrome (secondary to alcohol), colitis, rectal disorder, rhabdomyolysis, manic reaction (2 patients), convulsion, and depression (2 patients). Two patients reported more than 1 SAE possibly, probably, or definitely related to the study drug. Three patients (< 1% of the safety population) had SAEs considered possibly, probably, or definitely related to the study drug during the poststudy period (hyperthyroidism, 1 patient; convulsion, 2 patients). Serious AEs considered probably not or definitely not related to the study drug were reported by 31 patients (2% of the safety population) during the on-therapy period and 10 patients (< 1%) during the poststudy period. Five patients reported more than 1 SAE probably not or definitely not related to the study drug. No deaths occurred during the study and none were reported subsequently.
During the on-therapy or posttherapy periods, additional AEs judged by the study medical monitor to be of clinical importance were reported by 102 of 1,395 patients (7%) in the safety population. Posttherapy discontinuation symptoms were defined a priori to be an event of clinical importance, and 81 of the 102 patients reporting AEs that were determined to be of clinical importance reported withdrawal syndrome. Other AEs of clinical importance were chest pain, peripheral vascular disorder (cold feet), atrial fibrillation, hypertension, syncope, myocardial ischemia, parotid gland enlargement, prolactin increase (2 patients), leukopenia, ecchymosis, dizziness, extraocular palsy, visual field defect, urticaria, rash, urinary retention, breast secretion, and suicidal ideation (3 patients).
Laboratory assessments. Significant mean changes from open-label baseline values occurred at 1 or more scheduled evaluations for each of the following laboratory tests: creatinine, alkaline phosphatase, γ-glutamyl transpeptidase (GGT), aspartate aminotransferase/serum glutamic-oxaloacetic transaminase (AST/SGOT), alanine aminotransferase/serum glutamic-pyruvic transaminase (ALT/SGPT), total bilirubin, fasting total cholesterol, high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, and triglycerides. Significant mean changes from open-label baseline to the final evaluation (Table 3) were observed for alkaline phosphatase (+ 1.6 U/L; P < .001), GGT (+ 6.0 U/L; P < .001), total bilirubin (−0.042 mg/dL; P < .001), total cholesterol (+ 4.90 mg/dL; P < .001), HDL cholesterol (+ 1.95 mg/dL; P < .001), and triglycerides (+ 4.57 mg/dL; P < .001). Results of an analysis of change from the acute-phase baseline by double-blind treatment group were similar, but several differences were observed: patients assigned to the placebo group in the short-term lead-in studies had no statistically significant change from acute-phase baseline in GGT or HDL cholesterol at final evaluation after open-label desvenlafaxine treatment and had a statistically significant decrease in LDL cholesterol (−2.8 mg/dL; P < .01). Patients who had received short-term desvenlafaxine before enrolling in the extension study had statistically significant increases from acute-phase baseline in ALT/SGPT (+ 1.7 U/L; P < .001), AST/SGOT (+ 1.0 U/L; P < .001), and LDL cholesterol (+ 5.1 mg/dL; P < .001). None of the 3 groups had a statistically significant change from acute-phase baseline in triglyceride levels.
Click figure to enlarge
A total of 524 of 1,195 patients (44%) in the safety population who had laboratory assessments at least once during the on-therapy period had laboratory values of potential clinical importance based on FDA- or sponsor-defined criteria. Laboratory findings of clinical importance were determined by the study medical monitor after review of the baseline values and medical profile of each patient with potentially clinically important laboratory results. Clinically important changes in laboratory results were determined for 42 of 1,395 patients (3%) during the open-label period and included elevated cholesterol (n = 17), elevated liver function test (n = 9), anemia (n = 6), elevated triglycerides (n = 3), hematuria (n = 3), elevated thyroid function (n = 1), elevated serum glucose (n = 1), decreased white blood cells (n = 1), and Howell-Jolly bodies (n = 1).
Vital signs and weight. Significant mean increases from open-label baseline to the final evaluation (Table 3) were observed for supine systolic blood pressure (+ 2.58 mm Hg; P < .001), supine diastolic blood pressure (+ 1.20 mm Hg; P < .001), and pulse rate (+ 3.26 bpm; P < .001). Significant increases in those measures were also observed from the acute-phase baseline in each of the double-blind treatment groups, with numerically smaller changes for the patients previously assigned to placebo compared with those who had received acute-phase desvenlafaxine or venlafaxine ER (data not shown). At final evaluation, patients had a mean weight gain of 1.25 kg from open-label baseline (P < .001). The mean change from acute-phase baseline to final open-label evaluation was smaller because patients assigned to desvenlafaxine and venlafaxine ER groups had statistically significant weight loss during the acute-phase lead-in studies. Patients previously assigned to double-blind desvenlafaxine or venlafaxine ER lost 1.22 kg (P < .001) and 0.54 kg (P < .01), respectively, from acute-phase baseline to open-label baseline (placebo, + 0.04 kg [NS]), and gained 0.61 kg (P < .001), and 0.99 kg (P < .01), respectively, from acute-phase baseline to final evaluation (placebo, + 0.39 kg [P < .05]). The difference between treatment groups was statistically significant at the end of the acute phase (week 8: P < .001), but not at final evaluation. During the extension study, a small decrease from baseline in mean weight observed during the first month of open-label treatment (statistically significant at week 1 only [−0.07 kg; P < .05]) was followed by increasing weight. Mean weight was statistically significantly greater than open-label baseline from month 2 through month 10 (P < .001).
A total of 1,353 patients in the safety population had weight or vital sign measurements at least once during the on-therapy period. Of those, 456 (34%) had potentially clinically important results; 46 patients (3%) were determined to have results of clinical importance. Clinically important weight and vital sign findings included hypertension (n = 6), hypotension (n = 2), postural hypotension (n = 2), weight gain (n = 30), and weight loss (n = 6). Clinically significant weight gain ranged from 5.0 kg (7.9% of baseline) to 22.7 kg (30.1%); clinically significant weight loss ranged from 9.2 kg (13.5% of baseline) to 22.0 kg (23.7%).
Electrocardiogram. Mean change from open-label baseline in ECG measurements is shown in Table 3. Significant mean decreases from baseline were observed at on-therapy final evaluation for PR interval (−3.17 ms; P < .001), RR interval (−40.57 ms; P < .001), uncorrected QT interval (−5.17 ms; P < .001), and QT interval with correction based on the population correction factor (−6.43 ms; P < .001). Significant mean increases from baseline were observed for heart rate (3.47 bpm; P < .001), QRS interval (0.78 ms; P < .001), and QT interval with corrections using the Bazett formula (QTcB; 4.24 ms; P < .001) and using the Fridericia formula (QTcF; 0.97 ms; P < .05). Mean changes from acute-phase baseline to final evaluation were also statistically significant for the 3 previously assigned acute-phase treatment groups (data not shown).
A total of 238 of the 1,161 patients (21%) in the safety population who had at least 1 ECG recording during the on-therapy period had changes of potential clinical importance. Clinically important ECG findings were observed in 4 patients (< 1%): QRS prolongation (n = 2), left anterior hemiblock (n = 1), and increased QTcF and QTcB (n = 1).
Efficacy
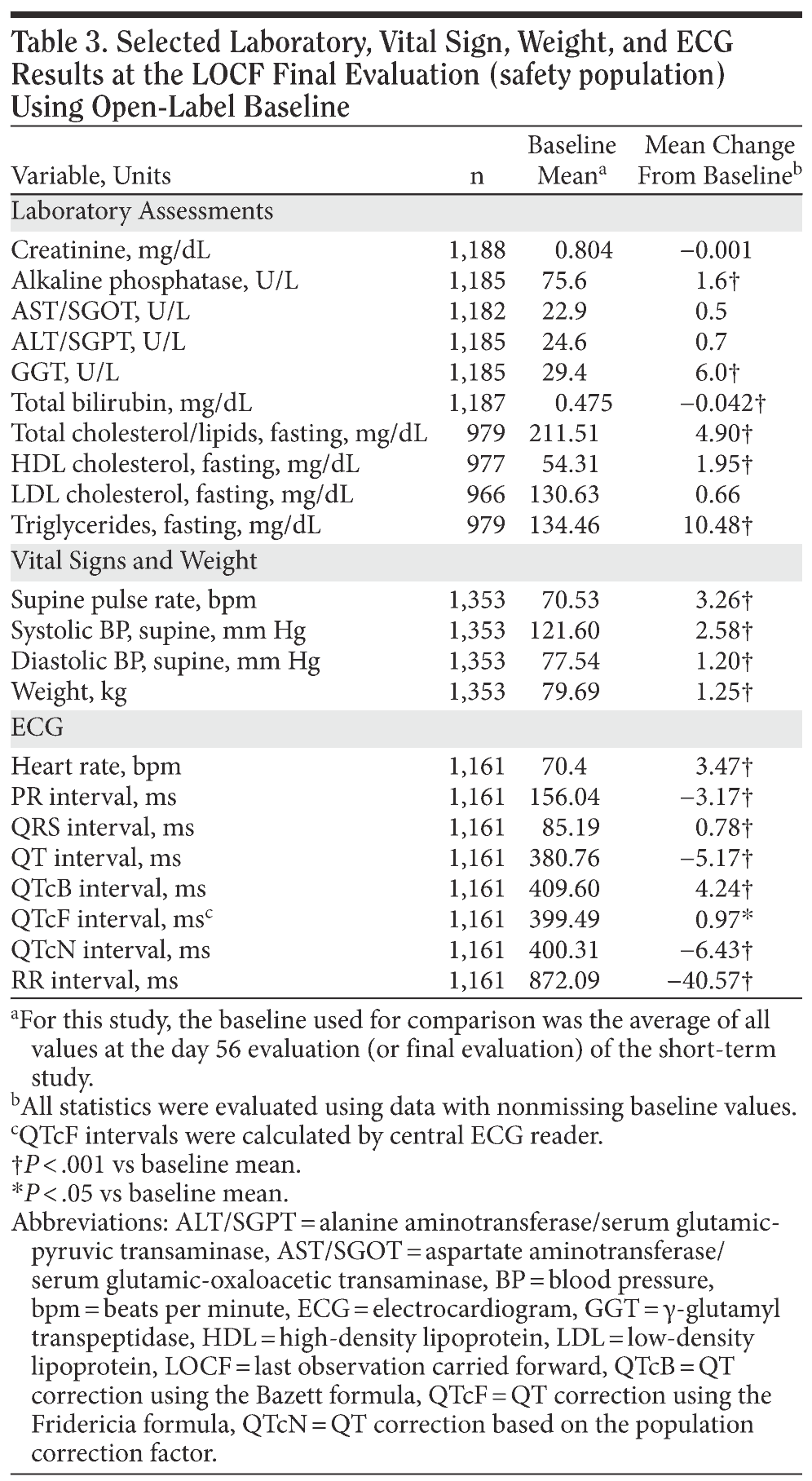
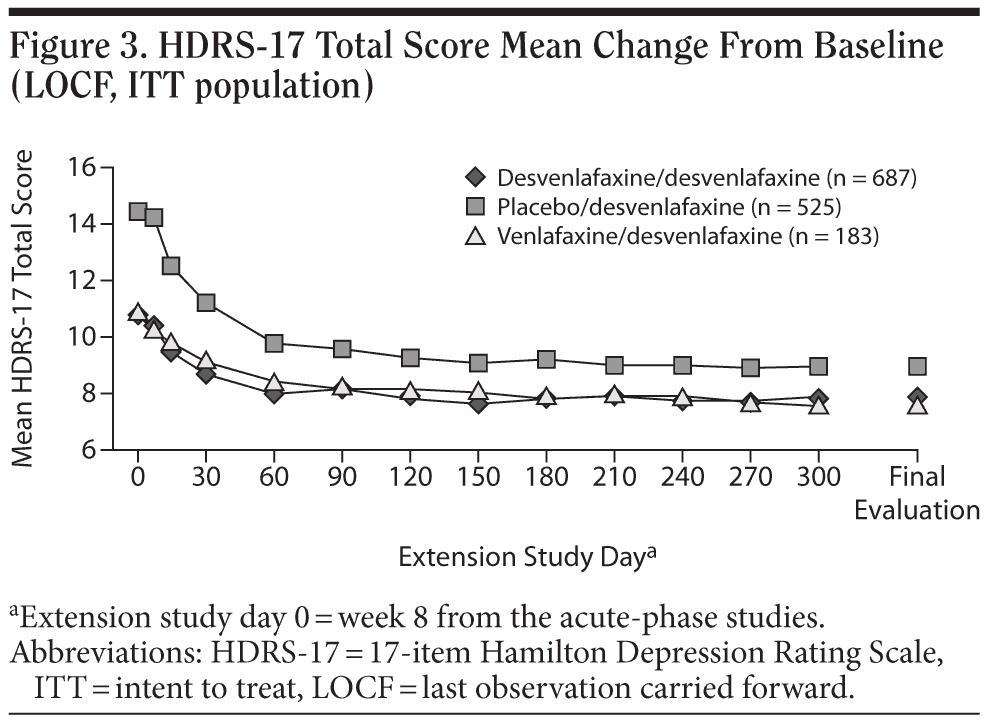
For the entire ITT population, the mean change in HDRS-17 total score from open-label baseline to the final evaluation (LOCF) was −3.9 ± 0.283. Patients who had received double-blind treatment with desvenlafaxine or venlafaxine ER in the short-term lead-in studies achieved small mean decreases from open-label baseline over the first 2 months of open-label treatment with desvenlafaxine, which were maintained throughout the rest of the 10-month study (mean open-label baseline scores: 10.8 ± 6.9 and 10.8 ± 7.1, respectively; change from open-label baseline to final evaluation, −3.0 ± 0.4 and −3.2 ± 0.7, respectively; Figure 3). Patients randomly assigned to placebo in the short-term studies, who had a higher mean open-label baseline score (14.4 ± 7.5) compared with patients assigned to desvenlafaxine or venlafaxine ER in the contributing studies, achieved a numerically larger mean decrease in HDRS-17 total scores (−5.5 ± 0.5) at final evaluation. Overall, remission rates improved from 32% at the open-label baseline (after 8-week double-blind treatment) to 58% at final evaluation. The proportions of patients who had achieved remission at the open-label baseline were 38% for patients who received short-term desvenlafaxine or venlafaxine ER treatment and 22% for patients who received placebo. Remission rates at the final evaluation (LOCF) of the open-label desvenlafaxine treatment period were 60%, 63%, and 54% for patients previously assigned to acute-phase desvenlafaxine, venlafaxine, and placebo, respectively (Figure 4).
Click figure to enlarge
Click figure to enlarge
DISCUSSION
This study reports the results of long-term, open-label treatment with flexible-dose desvenlafaxine in a large cohort of patients with MDD who had previously completed 8-week double-blind treatment with desvenlafaxine, venlafaxine ER, or placebo. The overall results indicate that desvenlafaxine has a long-term safety profile similar to that observed in short-term desvenlafaxine MDD studies22 and desvenlafaxine treatment up to 9 months.35 The results suggest that desvenlafaxine, studied here at 4 to 8 times the recommended dose, can be safely administered for up to 12 months.
The rate of discontinuations due to AEs in this study was 21%, likely due to both the duration of the study and the dose range used. Similar rates were observed in a 9-month MDD relapse-prevention study using desvenlafaxine 200 to 400 mg/d: 14% of patients treated with desvenlafaxine and 18% of patients treated with placebo cited AEs as the primary or secondary reason for study discontinuation during a 6-month, double-blind relapse-prevention phase.35 The recommended desvenlafaxine dose is 50 mg/d, but a higher dose range was used in the current study. In short-term studies, the 50-mg/d dose was associated with better tolerability compared with higher doses. Rates of discontinuation due to AEs were similar for the 50-mg/d desvenlafaxine dose and placebo (both ~4%) in an integrated analysis of safety and tolerability in 5 fixed-dose desvenlafaxine trials for MDD; higher dose groups (up to 400 mg/d) had rates as high as 18%.22 Rates of discontinuation due to AEs would therefore very likely be lower over the long term with the recommended 50-mg/d desvenlafaxine dose. Rates of discontinuation due to AEs observed in long-term treatment (6 months to 2 years) of MDD with other SNRIs (venlafaxine, duloxetine, and milnacipran) range from 3% to 21%.36-46
Treatment-emergent AEs reported by 10% or more patients in this study—headache, nausea, hyperhidrosis, dizziness, dry mouth, insomnia, upper respiratory infection, nasopharyngitis, and fatigue—are consistent with those reported in double-blind, placebo-controlled studies of desvenlafaxine 50 to 400 mg/d, particularly for higher dose groups in those studies.17-20 Adverse events associated with desvenlafaxine treatment are similar to those reported in the literature for long-term treatment with other SNRIs.36 Serious adverse events and noteworthy AEs were reported by < 5% of patients in the safety population and no deaths were reported during the study. Laboratory findings of note occurred for liver function, lipids, and urine protein. Few individual patients, however, had values that were considered clinically important by the medical monitor.
Adverse events that are of particular concern during long-term antidepressant treatment and can reduce treatment adherence include sexual dysfunction and weight gain.47-52 Rates of AEs related to sexual function were low in this study. However, no formal sexual functioning scale was administered, and spontaneous reports may underestimate the prevalence of treatment-emergent sexual dysfunction.53 Mean weight showed a pattern of initial decrease from baseline early in treatment, followed by a statistically significant mean increase from baseline of 1.25 kg at the final evaluation. This pattern of weight change is consistent with that reported in long-term studies with selective serotonin-reuptake inhibitors and with duloxetine.40,41,54 A similar pattern was also observed in desvenlafaxine-treated patients enrolled in the 6-month, double-blind phase of a placebo-controlled relapse-prevention study.35 In that study, however, both desvenlafaxine (200 to 400 mg/d; n = 190) and placebo groups (n = 185) had small mean decreases in weight followed by small but significant increases (< 1 kg) from baseline to final evaluation; there was no statistically significant difference between treatment groups.55 That result suggested that the pattern of weight increase was not specific to the study drug but rather may be due to improvement of MDD symptoms.56 In the current study, clinically important weight increases and decreases were seen in 2% and < 1%, respectively, of patients in the safety population.
Treatment with venlafaxine, the parent compound of desvenlafaxine, has been associated with dose-related changes in blood pressure.57 In a pooled analysis of 3,744 patients enrolled in short-term MDD trials, 9.1% of patients treated with greater than 300 mg/d of venlafaxine had sustained supine diastolic blood pressure (≥ 90 mm Hg). In the current study, statistically significant mean increases in supine systolic and diastolic blood pressure from baseline were observed with desvenlafaxine treatment at most time points. However, clinically important hypertension and postural hypotension were each identified in < 1% of patients in the safety population. Electrocardiogram changes were related to increased heart rate and otherwise were not clinically important.
The efficacy of desvenlafaxine was maintained throughout the 10-month duration of this study. Although interpretation of efficacy results is limited by the open-label study design and lack of a placebo control, there was a mean decrease in HDRS-17 total score from open-label baseline to final evaluation, with the greatest improvement observed during the first 2 months of open-label treatment. Overall, 58% of patients had achieved HDRS-17 remission at final evaluation, an increase from 32% at open-label week 1.
Strengths of the current study include the documentation of long-term safety and tolerability outcomes among outpatients with MDD, and the flexible-dose treatment, which is typical of clinical practice and permits optimal dose adjustment for each patient. The study visit structure was more similar to typical practice than the intensity of the visit structure in acute short-term studies. The study was limited by the desvenlafaxine dose range used (200 to 400 mg/d), which was 4 to 8 times higher than the current recommended therapeutic dose. Based on findings from short-term studies of desvenlafaxine for MDD, the recommended 50-mg/d dose is likely to be better tolerated during long-term treatment compared with the higher doses assessed here.22 The lack of a placebo control limits the conclusions that can be drawn, and the multiple exclusion criteria may limit generalization of these results to MDD patients with clinically significant comorbid conditions.
CONCLUSIONS
In this long-term, open-label, flexible-dose study, desvenlafaxine had a safety profile similar to that observed in short-term (up to 8 weeks) desvenlafaxine MDD studies with similar doses. Rates of AEs and discontinuations due to AEs observed in patients receiving desvenlafaxine doses from 200 to 400 mg/d in this study are likely to be high compared with those expected for the recommended dose of 50 mg/d. No new safety findings were observed in this study. Symptoms of depression improved during long-term desvenlafaxine treatment, and improvement was sustained through the final evaluation. Desvenlafaxine is safe and effective when administered for up to 12 months.
Drug names: desvenlafaxine (Pristiq), duloxetine (Cymbalta), milnacipran (Savella), venlafaxine (Effexor and others).
Author affiliations: Wyeth Pharmaceuticals France, Paris (Drs Tourian and Pitrosky); and Pfizer, Collegeville, Pennsylvania (Drs Padmanabhan and Rosas).
Potential conflicts of interest: Drs Tourian and Pitrosky are full-time employees of Wyeth, a Company of the Pfizer Group, Division Wyeth Research, Paris, France. Drs Padmanabhan and Rosas are full-time employees of Pfizer Inc, formerly Wyeth Research, Collegeville, Pennsylvania. Dr Tourian is a stock shareholder in Pfizer.
Funding/support: This study was sponsored by Wyeth, which was acquired by Pfizer Inc in October 2009. Medical writing support for this manuscript was provided by Kathleen Dorries, PhD, and medical editing support by Jennifer Karpinski, BA, both of Embryon, LLC, a Division of Advanced Health Media, LLC (formerly Medesta Publications Group, a Business of Advogent) and were funded by Pfizer Inc.
Acknowledgment: The authors thank the Study 303 investigators for their valuable contribution.
REFERENCES
1. Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet. 1997;349(9063):1436-1442. PubMed doi:10.1016/S0140-6736(96)07495-8
2. Ust×¼n TB, Ayuso-Mateos JL, Chatterji S, et al. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184(5):386-392. PubMed doi:10.1192/bjp.184.5.386
3. Greenberg PE, Kessler RC, Birnbaum HG, et al. The economic burden of depression in the United States: how did it change between 1990 and 2000? J Clin Psychiatry. 2003;64(12):1465-1475. PubMed doi:10.4088/JCP.v64n1211
4. Hirschfeld RM, Montgomery SA, Keller MB, et al. Social functioning in depression: a review. J Clin Psychiatry. 2000;61(4):268-275. PubMed
5. Moussavi S, Chatterji S, Verdes E, et al. Depression, chronic diseases, and decrements in health: results from the World Health Surveys. Lancet. 2007;370(9590):851-858. PubMed doi:10.1016/S0140-6736(07)61415-9
6. Vuorilehto M, Melartin T, Isometsä E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med. 2005;35(5):673-682. PubMed doi:10.1017/S0033291704003770
7. Vuorilehto MS, Melartin TK, Isometsä ET. Course and outcome of depressive disorders in primary care: a prospective 18-month study. Psychol Med. 2009;39(10):1697-1707. PubMed doi:10.1017/S0033291709005182
8. Yiend J, Paykel E, Merritt R, et al. Long term outcome of primary care depression. J Affect Disord. 2009;118(1-3):79-86. PubMed doi:10.1016/j.jad.2009.01.026
9. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry. 2000;157(4 suppl):1-45. PubMed
10. National Collaborating Centre for Mental Health. Depression: management of depression in primary and secondary care: guideline 23; http://www.scamfyc.org/documentos/depresion%20NICE.pdf. Accessed September 17, 2010. Updated December 2004.
11. Geddes JR, Carney SM, Davies C, et al. Relapse prevention with antidepressant drug treatment in depressive disorders: a systematic review. Lancet. 2003;361(9358):653-661. PubMed doi:10.1016/S0140-6736(03)12599-8
12. Deecher DC, Beyer CE, Johnston G, et al. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther. 2006;318(2):657-665. PubMed doi:10.1124/jpet.106.103382
13. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc; 2008.
14. Patroneva A, Connolly SM, Fatato P, et al. An assessment of drug-drug interactions: the effect of desvenlafaxine and duloxetine on the pharmacokinetics of the CYP2D6 probe desipramine in healthy subjects. Drug Metab Dispos. 2008;36(12):2484-2491. PubMed doi:10.1124/dmd.108.021527
15. Nichols AI, Fatato P, Shenouda M, et al. The effects of desvenlafaxine and paroxetine on the pharmacokinetics of the cytochrome P450 2D6 substrate desipramine in healthy adults. J Clin Pharmacol. 2009;49(2):219-228. PubMed doi:10.1177/0091270008326716
16. Oganesian A, Shilling AD, Young-Sciame R, et al. Desvenlafaxine and venlafaxine exert minimal in vitro inhibition of human cytochrome p450 and p-glycoprotein activities. Psychopharmacol Bull. 2009;42(2):47-63. PubMed
17. DeMartinis NA, Yeung PP, Entsuah R, et al. A double-blind, placebo-controlled study of the efficacy and safety of desvenlafaxine succinate in the treatment of major depressive disorder. J Clin Psychiatry. 2007;68(5):677-688. PubMed doi:10.4088/JCP.v68n0504
18. Septien-Velez L, Pitrosky B, Padmanabhan SK, et al. A randomized, double-blind, placebo-controlled trial of desvenlafaxine succinate in the treatment of major depressive disorder. Int Clin Psychopharmacol. 2007;22(6):338-347. PubMed doi:10.1097/YIC.0b013e3281e2c84b
19. Boyer P, Montgomery S, Lepola U, et al. Efficacy, safety, and tolerability of fixed-dose desvenlafaxine 50 and 100 mg/day for major depressive disorder in a placebo-controlled trial. Int Clin Psychopharmacol. 2008;23(5):243-253. PubMed doi:10.1097/YIC.0b013e32830cebed
20. Liebowitz MR, Manley AL, Padmanabhan SK, et al. Efficacy, safety, and tolerability of desvenlafaxine 50 mg/day and 100 mg/day in outpatients with major depressive disorder. Curr Med Res Opin. 2008;24(7):1877-1890. PubMed doi:10.1185/03007990802161923
21. Thase ME, Kornstein SG, Germain J-M, et al. An integrated analysis of the efficacy of desvenlafaxine compared with placebo in patients with major depressive disorder. CNS Spectr. 2009;14(3):144-154. PubMed
22. Clayton AH, Kornstein SG, Rosas G, et al. An integrated analysis of the safety and tolerability of desvenlafaxine compared with placebo in the treatment of major depressive disorder. CNS Spectr. 2009;14(4):183-195. PubMed
23. Liebowitz MR, Yeung PP, Entsuah R. A randomized, double-blind, placebo-controlled trial of desvenlafaxine succinate in adult outpatients with major depressive disorder. J Clin Psychiatry. 2007;68(11):1663-1672. PubMed doi:10.4088/JCP.v68n1105
24. Lieberman DZ, Montgomery SA, Tourian KA, et al. A pooled analysis of two placebo-controlled trials of desvenlafaxine in major depressive disorder. Int Clin Psychopharmacol. 2008;23(4):188-197. PubMed doi:10.1097/YIC.0b013e32830263de
25. Feiger AD, Tourian KA, Rosas GR, et al. A placebo-controlled study evaluating the efficacy and safety of flexible-dose desvenlafaxine treatment in outpatients with major depressive disorder. CNS Spectr. 2009;14(1):41-50. PubMed
26. Food and Drug Administration. FDA Code of Federal Register 21CFR, part 50. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?cfrpart=50&showfr=1. Accessed November 21, 2010.
27. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). E6 R1-European Medicines Agency. Guideline for Good Clinical Practice, 2002, 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002874.pdf. Accessed December 10, 2010.
28. National Institutes of Health. World Medical Association Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. http://ohsr.od.nih.gov/guidelines/helsinki.html. Accessed November 20, 2010.
29. Food and Drug Administration. FDA Good Clinical Practice Guidelines. http://www.fda.gov/ScienceResearch/SpecialTopics/RunningClinicalTrials/GuidancesInformationSheetsandNotices/default.htm. Accessed December 10 2010.
30. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. Washington, DC: American Psychiatric Association; 1994.
31. Rosenbaum JF, Fava M, Hoog SL, et al. Selective serotonin reuptake inhibitor discontinuation syndrome: a randomized clinical trial. Biol Psychiatry. 1998;44(2):77-87.1016/S0006-3223(98)00126-7 PubMed
32. Medical Dictionary for Regulatory Activities (MedDRA), 10th ed (Wyeth-modified). http://www.meddramsso.com/. Accessed December 10, 2010.
33. National Technical Information Service. Coding Symbols for a Thesaurus of Adverse Reaction Terms (COSTART; Wyeth-modified). 4th ed. Springfield, VA: National Technical Information Service; 1992.
34. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56-62. PubMed doi:10.1136/jnnp.23.1.56
35. Rickels K, Montgomery SA, Tourian KA, et al. Desvenlafaxine for the prevention of relapse in major depressive disorder: results of a randomized trial. J Clin Psychopharmacol. 2010;30(1):18-24. PubMed doi:10.1097/JCP.0b013e3181c94c4d
36. Kocsis JH, Thase ME, Trivedi MH, et al. Prevention of recurrent episodes of depression with venlafaxine ER in a 1-year maintenance phase from the PREVENT Study. J Clin Psychiatry. 2007;68(7):1014-1023. PubMed doi:10.4088/JCP.v68n0706
37. Kornstein SG, Dunner DL, Meyers AL, et al. A randomized, double-blind study of increasing or maintaining duloxetine dose in patients without remission of major depressive disorder after initial duloxetine therapy. J Clin Psychiatry. 2008;69(9):1383-1392. PubMed doi:10.4088/JCP.v69n0905
38. Entsuah AR, Rudolph RL, Hackett D, et al. Efficacy of venlafaxine and placebo during long-term treatment of depression: a pooled analysis of relapse rates. Int Clin Psychopharmacol. 1996;11(2):137-145. PubMed
39. Montgomery SA, Entsuah R, Hackett D, et al; Venlafaxine 335 Study Group. Venlafaxine versus placebo in the preventive treatment of recurrent major depression. J Clin Psychiatry. 2004;65(3):328-336. PubMed doi:10.4088/JCP.v65n0307
40. Dunner DL, Wilson M, Fava M, et al. Long-term tolerability and effectiveness of duloxetine in the treatment of major depressive disorder. Depress Anxiety. 2008;25(5):E1-E8. PubMed doi:10.1002/da.20339
41. Raskin J, Goldstein DJ, Mallinckrodt CH, et al. Duloxetine in the long-term treatment of major depressive disorder. J Clin Psychiatry. 2003;64(10):1237-1244. PubMed doi:10.4088/JCP.v64n1015
42. Leinonen E, Lepola U, Koponen H, et al. Long-term efficacy and safety of milnacipran compared to clomipramine in patients with major depression. Acta Psychiatr Scand. 1997;96(6):497-504. PubMed doi:10.1111/j.1600-0447.1997.tb09953.x
43. Schweitzer I, Burrows G, Tuckwell V, et al. Sustained response to open-label venlafaxine in drug-resistant major depression. J Clin Psychopharmacol. 2001;21(2):185-189. PubMed doi:10.1097/00004714-200104000-00010
44. Shrivastava RK, Cohn C, Crowder J, et al. Long-term safety and clinical acceptability of venlafaxine and imipramine in outpatients with major depression. J Clin Psychopharmacol. 1994;14(5):322-329. PubMed
45. Steen A, den Boer JA. A double-blind six months comparative study of milnacipran and clomipramine in major depressive disorder. Int Clin Psychopharmacol. 1997;12(5):269-281. PubMed doi:10.1097/00004850-199709000-00004
46. Wade A, Gembert K, Florea I. A comparative study of the efficacy of acute and continuation treatment with escitalopram versus duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2007;23(7):1605-1614. PubMed doi:10.1185/030079907X210732
47. Cassano P, Fava M. Tolerability issues during long-term treatment with antidepressants. Ann Clin Psychiatry. 2004;16(1):15-25. PubMed
48. Montgomery SA, Baldwin DS, Riley A. Antidepressant medications: a review of the evidence for drug-induced sexual dysfunction. J Affect Disord. 2002;69(1-3):119-140. PubMed doi:10.1016/S0165-0327(01)00313-5
49. Werneke U, Northey S, Bhugra D. Antidepressants and sexual dysfunction. Acta Psychiatr Scand. 2006;114(6):384-397. PubMed doi:10.1111/j.1600-0447.2006.00890.x
50. Berken GH, Weinstein DO, Stern WC. Weight gain: a side-effect of tricyclic antidepressants. J Affect Disord. 1984;7(2):133-138. PubMed doi:10.1016/0165-0327(84)90031-4
51. Zajecka JM. Clinical issues in long-term treatment with antidepressants. J Clin Psychiatry. 2000;61(suppl 2):20-25. PubMed
52. Burra TA, Chen E, McIntyre RS, et al. Predictors of self-reported antidepressant adherence. Behav Med. 2007;32(4):127-134. PubMed doi:10.3200/BMED.32.4.127-134
53. Montejo-Gonzסlez AL, Llorca G, Izquierdo JA, et al. SSRI-induced sexual dysfunction: fluoxetine, paroxetine, sertraline, and fluvoxamine in a prospective, multicenter, and descriptive clinical study of 344 patients. J Sex Marital Ther. 1997;23(3):176-194. PubMed
54. Sussman N, Ginsberg DL, Bikoff J. Effects of nefazodone on body weight: a pooled analysis of selective serotonin reuptake inhibitor- and imipramine-controlled trials. J Clin Psychiatry. 2001;62(4):256-260. PubMed doi:10.4088/JCP.v62n0407
55. Tourian KA, Leurent C, Graepel J, et al. Desvenlafaxine and weight change in major depressive disorder. Prim Care Companion J Clin Psychiatry. 2010;12(1):e1-e8. PubMed
56. Benazzi F. Weight gain in depression remitted with antidepressants: pharmacological or recovery effect? Psychother Psychosom. 1998;67(4-5):271-274. PubMed doi:10.1159/000012291
57. Thase ME. Effects of venlafaxine on blood pressure: a meta-analysis of original data from 3744 depressed patients. J Clin Psychiatry. 1998;59(10):502-508. PubMed


