Objective: The avoidance of adverse drug-drug interactions (DDIs) is a high priority in terms of both the US Food and Drug Administration (FDA) and the individual prescriber. With this perspective in mind, this article illustrates the process for assessing the risk of a drug (example here being desvenlafaxine) causing or being the victim of DDIs, in accordance with FDA guidance.
Data Sources/Study Selection: DDI studies for the serotonin-norepinephrine reuptake inhibitor desvenlafaxine conducted by the sponsor and published since 2009 are used as examples of the systematic way that the FDA requires drug developers to assess whether their new drug is either capable of causing clinically meaningful DDIs or being the victim of such DDIs. In total, 8 open-label studies tested the effects of steady-state treatment with desvenlafaxine (50-400 mg/d) on the pharmacokinetics of cytochrome (CYP) 2D6 and/or CYP 3A4 substrate drugs, or the effect of CYP 3A4 inhibition on desvenlafaxine pharmacokinetics. The potential for DDIs mediated by the P-glycoprotein (P-gp) transporter was assessed in in vitro studies using Caco-2 monolayers.
Data Extraction: Changes in area under the plasma concentration-time curve (AUC; CYP studies) and efflux (P-gp studies) were reviewed for potential DDIs in accordance with FDA criteria.
Results: Desvenlafaxine coadministration had minimal effect on CYP 2D6 and/or 3A4 substrates per FDA criteria. Changes in AUC indicated either no interaction (90% confidence intervals for the ratio of AUC geometric least-squares means [GM] within 80%-125%) or weak inhibition (AUC GM ratio 125% to < 200%). Coadministration with ketoconazole resulted in a weak interaction with desvenlafaxine (AUC GM ratio of 143%). Desvenlafaxine was not a substrate (efflux ratio < 2) or inhibitor (50% inhibitory drug concentration values > 250 μM) of P-gp.
Conclusions: A 2-step process based on FDA guidance can be used first to determine whether a pharmacokinetically mediated interaction occurs and then to assess the potential clinical significance of the DDI. In the case of the drug tested in this series of studies, the potential for clinically meaningful DDIs mediated by CYP 2D6, CYP 3A4, or P-gp was found to be low.
How the Probability and Potential Clinical Significance of Pharmacokinetically Mediated Drug-Drug Interactions Are Assessed in Drug Development: Desvenlafaxine as an Example
ABSTRACT
Objective: The avoidance of adverse drug-drug interactions (DDIs) is a high priority in terms of both the US Food and Drug Administration (FDA) and the individual prescriber. With this perspective in mind, this article illustrates the process for assessing the risk of a drug (example here being desvenlafaxine) causing or being the victim of DDIs, in accordance with FDA guidance.
Data Sources/Study Selection: DDI studies for the serotonin-norepinephrine reuptake inhibitor desvenlafaxine conducted by the sponsor and published since 2009 are used as examples of the systematic way that the FDA requires drug developers to assess whether their new drug is either capable of causing clinically meaningful DDIs or being the victim of such DDIs. In total, 8 open-label studies tested the effects of steady-state treatment with desvenlafaxine (50–400 mg/d) on the pharmacokinetics of cytochrome (CYP) 2D6 and/or CYP 3A4 substrate drugs, or the effect of CYP 3A4 inhibition on desvenlafaxine pharmacokinetics. The potential for DDIs mediated by the P-glycoprotein (P-gp) transporter was assessed in in vitro studies using Caco-2 monolayers.
Data Extraction: Changes in area under the plasma concentration-time curve (AUC; CYP studies) and efflux (P-gp studies) were reviewed for potential DDIs in accordance with FDA criteria.
Results: Desvenlafaxine coadministration had minimal effect on CYP 2D6 and/or 3A4 substrates per FDA criteria. Changes in AUC indicated either no interaction (90% confidence intervals for the ratio of AUC geometric least-squares means [GM] within 80%–125%) or weak inhibition (AUC GM ratio 125% to < 200%). Coadministration with ketoconazole resulted in a weak interaction with desvenlafaxine (AUC GM ratio of 143%). Desvenlafaxine was not a substrate (efflux ratio < 2) or inhibitor (50% inhibitory drug concentration values > 250 μM) of P-gp.
Conclusions: A 2-step process based on FDA guidance can be used first to determine whether a pharmacokinetically mediated interaction occurs and then to assess the potential clinical significance of the DDI. In the case of the drug tested in this series of studies, the potential for clinically meaningful DDIs mediated by CYP 2D6, CYP 3A4, or P-gp was found to be low.
Prim Care Companion CNS Disord 2015;17(2):doi:10.4088/PCC.14r01710
© Copyright 2015 Physicians Postgraduate Press, Inc.
Submitted: August 13, 2014; accepted October 29, 2014.
Published online: March 19, 2015.
Corresponding author: Matthew Macaluso, DO, Department of Psychiatry and Behavioral Sciences, University of Kansas School of Medicine–Wichita, 1010 North Kansas, Wichita, KS 67214-3199 ([email protected]).
The US Food and Drug Administration (FDA) states that a new drug application should include an assessment of the potential for drug-drug interactions (DDIs) and outlines the process for conducting DDI studies and interpreting the study results.1 Practicing clinicians should both understand the importance of assessing the risk for clinically significant DDIs in new drugs and be familiar with the process by which that assessment is made. The number of patients prescribed antidepressant drugs,2 together with the prevalence and complexity of multiple medication use in that patient population,3 means that a substantial proportion of patients seen by primary care practitioners and specialists are likely to be at risk for DDI.
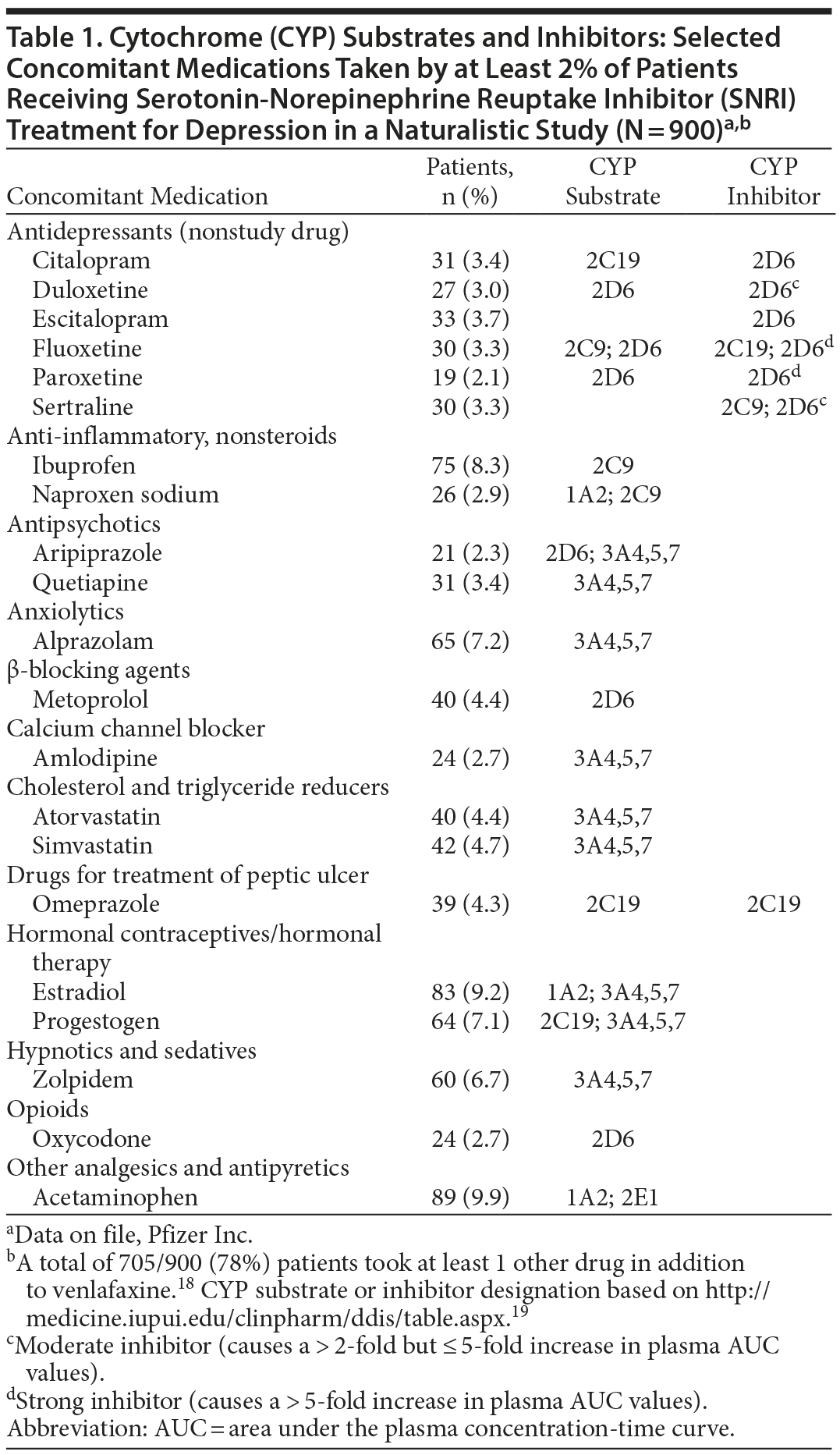
Major depressive disorder (MDD) is often difficult to treat to remission,4,5 and treatment strategies include augmentation of antidepressants with other drugs.4,6,7 Patients with MDD also have high rates of comorbidity with mental disorders8 and general medical conditions.9–16 In general, the greater the number of medications prescribed, the higher the risk to the patient.17,18 Consequently, patients treated with antidepressants are significantly more likely to receive greater numbers of medications compared with patients taking nonantidepressant drugs3 and are at increased risk for DDI. A listing of some of the most common concomitant medications used by depressed patients receiving antidepressant treatment in real-world clinical practice18,19 reflects the multiple medication use associated with MDD (Table 1). Concomitant antidepressants (the use of > 1 antidepressant) were reported by 25.2% of the study population, cholesterol and triglyceride reducers by 16.1%, analgesics and antipyretics by 15.8%, nonsteroidal anti-inflammatory/antirheumatic products by 14.4%, anxiolytics by 12.6%, drugs for peptic ulcer by 12.6%, and antiepileptics by 11.2%. In all, 78% of these antidepressant-treated patients took concomitant medications.18
METHOD
Despite the importance of assessment for potential risk of DDI, many prescribers are not knowledgeable about this part of the drug development process and how it impacts their care of patients. The intent of this article is to explain this process, including both the design of the studies that are conducted and how the results are interpreted to determine whether an interaction occurs and, if so, how to judge whether it is likely to be clinically meaningful and to what degree. The process will be illustrated using the studies that were done for the recently approved antidepressant desvenlafaxine by examining the potential for DDIs mediated by the cytochrome P450 (CYP) and P-glycoprotein (P-gp) systems. Desvenlafaxine pharmacokinetic studies conducted by the sponsor (Pfizer) and published in the past 5 years (since 2009) are reviewed to demonstrate the systematic process for assessing the likelihood for clinically meaningful DDI with a new drug based on FDA guidance. Eight open-label studies published in 5 articles20–24 tested the effects of steady-state treatment with desvenlafaxine (50–400 mg/d) on the pharmacokinetics of CYP 2D6 and/or CYP 3A4 substrate drugs and the effect of CYP 3A4 inhibition on desvenlafaxine pharmacokinetics. The potential for DDIs mediated by the P-gp transporter was assessed in in vitro studies using Caco-2 monolayers.25 Changes in drug exposure (CYP studies) and efflux (P-gp studies) were reviewed for potential DDIs in accordance with FDA criteria.
PHARMACOKINETICALLY MEDIATED
DRUG-DRUG INTERACTIONS
Among the most extensively studied systems implicated in pharmacokinetically mediated DDIs are the CYP system and the transport protein system, particularly P-gp. The CYP family of enzymes comprises the principal phase 1 metabolic pathway for most clinically used drugs.26,27 The CYP enzymes responsible for the greatest percentage of oxidative metabolism of drugs in humans are CYP 3A4 (36% of substrate interactions), CYP 2C (25%), and CYP 2D6 (15%).28 Numerous antidepressant drugs, including tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), and atypical antidepressants, interact with CYP enzymes as substrates or inhibitors.19,27
The nonmetabolic efflux transporter P-gp is found in the gastrointestinal tract, hepatocytes, kidney, blood-brain barrier, and placenta,27,29,30 and its activity can affect bioavailability or brain levels of substrate drugs.27,30 Substrates and/or inhibitors of P-gp include statin drugs,31 human immunodeficiency virus protease inhibitors,32 sex-steroid hormones,33 calcium-channel blockers,34 anticancer drugs,34 and psychotropic drugs, including antidepressants.27,35 A number of antidepressants in the TCA, SSRI, and SNRI classes are known to interact with the P-gp transporter.27,35
Activity of CYP enzymes or the P-gp transporter can be inhibited or induced by coadministered drugs, altering exposure to the substrate drug and its metabolites.31,36,37 Differences in exposure to medications related to CYP or P-gp activity can potentially affect safety, tolerability, or efficacy.38–43 Two examples illustrate potential risks of DDIs with antidepressant drugs. In the first example, venlafaxine, a CYP 2D6 substrate,19 is the potential victim of DDI in depressed patients taking concomitant CYP 2D6 inducers or inhibitors.18,39 Individuals can be classified as CYP 2D6 poor, intermediate, extensive, or ultrarapid metabolizers based on their metabolism of CYP 2D6 substrate drugs.26 Concomitant use of CYP 2D6 substrates or inhibitors with venlafaxine is associated with phenoconversion from extensive or ultra metabolizers to the poor metabolizer phenotype,18 and in patients with MDD treated with venlafaxine, there was a robust statistically significant difference between venlafaxine and placebo in terms of responder and remitter rates in CYP 2D6 extensive metabolizer individuals, but not in CYP 2D6 poor metabolizer individuals.39 In a second example of DDI with an antidepressant drug, paroxetine, which is both a CYP 2D6 substrate and a strong CYP 2D6 inhibitor,19 is the perpetrator of DDI with tamoxifen in women treated for estrogen receptor–positive breast cancer.36,37,44,45 Tamoxifen is metabolized sequentially by CYP 3A4 and CYP 2D6 to the active metabolite endoxifen.46,47 In women treated with tamoxifen, both the CYP 2D6 poor metabolizer phenotype and the use of concomitant treatment with paroxetine are associated with reduced exposure to endoxifen,36,37 and coadministration of paroxetine with tamoxifen is associated with a significantly increased risk of mortality.44 These examples underscore that DDIs with antidepressant drugs could reduce the efficacy of the antidepressant drug or of a concomitant drug, or even the efficacy of both, in the case of a drug that is both a substrate and an inhibitor of CYP enzymes, such as paroxetine.
Like phase I metabolism, phase II glucuronidation is mediated by a family of isozymes, the uridine 5′-diphospho-glucuronosyltransferase (UGT) enzymes.48 The potential for DDI between a new drug and concomitant medications that alter UGT activity depends, in part, on the number of UGT isozymes involved in the metabolism of the new drug: if glucuronidation is mediated by multiple UGTs, inhibition of 1 would be unlikely to have a clinically significant effect on drug exposure.49,50 Therefore, the likelihood of DDI at the UGT pathway level can be assessed by determining the number of UGT isozymes that can catalyze glucuronidation of a new drug.
ASSESSING THE POTENTIAL FOR PHARMACOKINETICALLY MEDIATED
DRUG-DRUG INTERACTIONS
In the drug development process, early steps prior to the conduct of clinical trials include in vitro and animal studies, followed by first-in-human studies and pharmacokinetic analyses. One of the goals of preclinical assessment is to understand the new drug’s disposition (absorption, distribution, metabolism, and elimination) in order to identify mechanisms with the potential for interference with or by other drugs. The new drug’s metabolism is characterized, and potential sites for interaction with the metabolism of other drugs—as either the perpetrator or the victim of DDI—are selected as the focus of pharmacokinetic studies.
This information was determined for desvenlafaxine during its preclinical development and provided the guidance for which DDI studies should be conducted. Desvenlafaxine is primarily metabolized by UGT conjugation to desvenlafaxine-glucuronide; approximately 19% is excreted as the glucuronide metabolite.51 Less than 5% of desvenlafaxine is excreted as the oxidative metabolite N,O-didesmethylvenlafaxine, a product of the CYP 3A4 pathway; approximately 45% of desvenlafaxine is excreted unchanged.51,52 On the basis of this information, desvenlafaxine was believed to have a low risk of either causing or being the victim of DDI mediated via either CYP 2D6 or 3A4 pathways, in contrast to a number of other psychiatric medications.53 However, antidepressants are frequently used with drugs that are substrates, inhibitors, or inducers of those CYP enzymes (see Table 1).18 The glucuronidation pathway and transporter interactions were also recognized as possible sites of interaction for desvenlafaxine, and preclinical studies assessing the potential for DDIs involving UGT and P-gp are also reviewed.
Determination of Risk for Drug-Drug Interactions Mediated via CYP Enzymes: Design and Interpretation of Pharmacokinetic Studies
Once relevant CYP pathways have been identified, the potential for CYP-mediated interactions can be assessed in in vivo pharmacokinetic studies (Table 2). These studies generally enroll healthy volunteers and test the new drug at doses used in clinical practice in an open-label design.
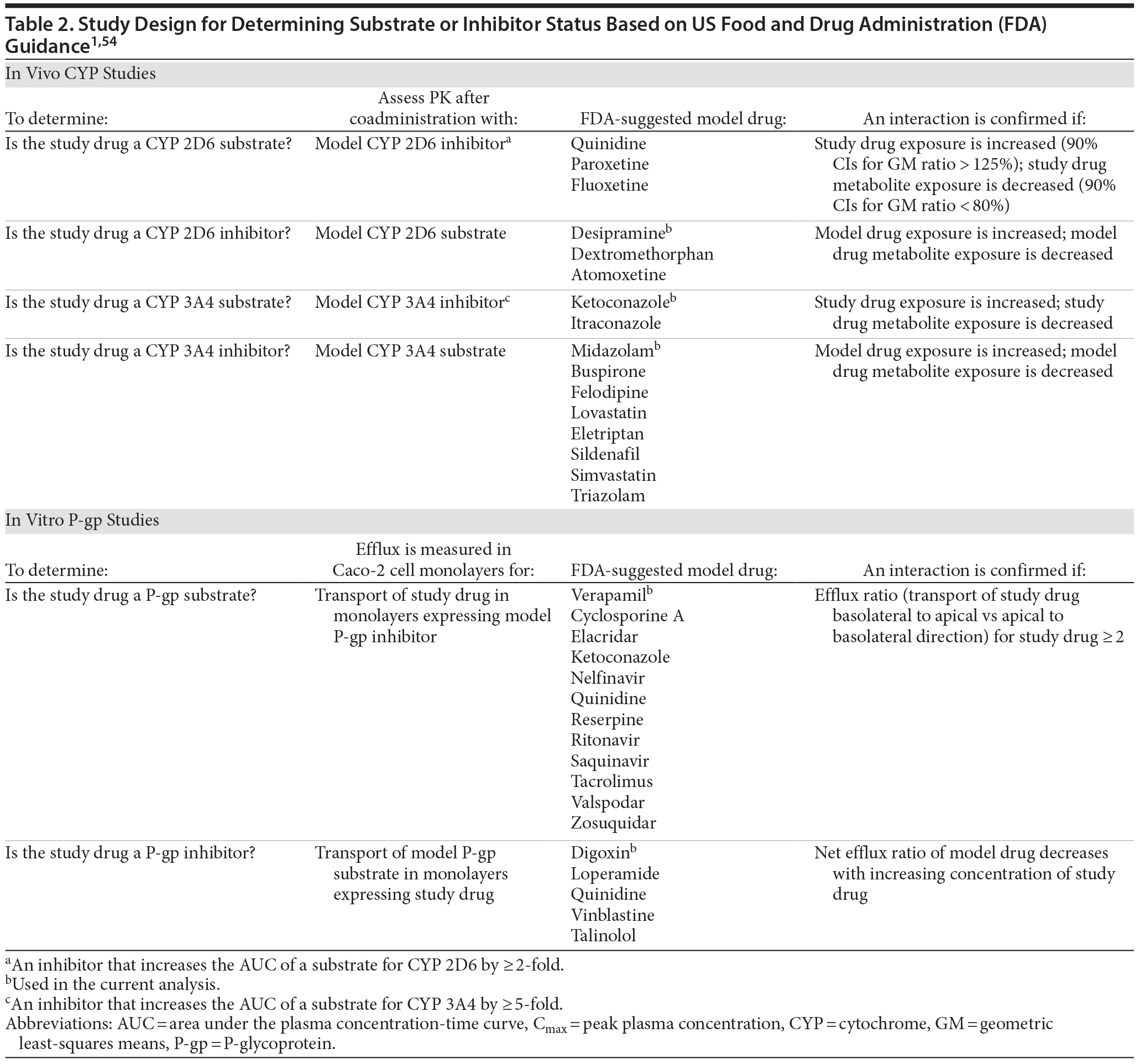
Pharmacokinetic DDI studies are designed to measure differences in exposure to the CYP substrate drug (quantified as area under the concentration-time curve [AUC] and peak plasma concentration [Cmax]) with and without a concomitant inhibitor or inducer of that enzyme. To test whether the new drug is a substrate, it is coadministered with a strong inhibitor of the enzyme, and to test whether the new drug is an inhibitor itself, it is administered with a model substrate drug that is known to show significant changes in concentration with coadministration of an inhibitor.1 Plasma concentrations of the substrate drug are measured for substrate alone and for substrate plus inhibitor or potential inhibitor either in separate arms of the study or using the more efficient within-subject crossover design. All of the desvenlafaxine studies used a crossover design in which subjects received a single dose of the substrate drug in phase 1, followed by once-daily dosing of the inhibitor and single dose of the substrate after steady-state plasma concentration of the inhibitor was reached in phase 2. If an active comparator is included in a DDI study, a randomized crossover design can be used to test half the subjects with the new drug first and half with the comparator first. The duration of each phase of the DDI study is dependent on pharmacokinetic and pharmacodynamic characteristics of the substrate and inhibitor drugs. Blood samples are collected at specified intervals throughout each study phase, and plasma substrate concentrations at each time point are used to calculate the AUC in the presence and absence of the inhibitor or potential inhibitor.
The magnitude of effect of the inhibitor on substrate drug exposure is estimated by comparing the AUC and Cmax of the substrate drug alone and in the presence of the interacting drug at steady state. The results are then analyzed in a 2-step process based on FDA guidance to determine whether an interaction has occurred and, if so, how likely the interaction is to be clinically significant. First, lack of an interaction between drugs can be concluded if the 90% confidence intervals (CIs) for the ratio of AUC geometric least-squares means (GM) fall wholly within a prespecified bioequivalent range of 80%–125%.1 Second, if an interaction is observed (ie, the GM ratio falls outside the bioequivalence range), the strength of the interaction can be described based on the magnitude of the GM ratio. FDA guidance defines weak inhibition based on an increase in substrate AUC of 125% to < 200%, moderate inhibition based on an increase in AUC of 200% but < 500%, and strong inhibition based on an increase in AUC of ≥ 5-fold (Figure 1).1 (For more information, see http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf.)

A series of single-center, open-label, single-dose, pharmacokinetic studies sponsored by Pfizer were designed to assess the risk for clinically meaningful DDIs involving desvenlafaxine and substrates or inhibitors of CYP 2D6 and/or 3A4 (Table 3). Desipramine20,21 and midazolam22 were used as the model substrate drugs because their metabolism is principally, if not wholly, dependent on a single CYP enzyme (CYP 2D6 and CYP 3A4, respectively). One desipramine study included the active comparator paroxetine in a 4-period, randomized crossover design.21 Two substrates, tamoxifen and aripiprazole, were used to assess the effect of desvenlafaxine on drugs metabolized via both the CYP 2D6 and CYP 3A4 pathways,23,24 either sequentially (tamoxifen) or concurrently (aripiprazole).36,55 The effect of CYP 3A4 inhibition on desvenlafaxine exposure was assessed using the known CYP 3A4 inhibitor ketoconazole.22,56
Desvenlafaxine was assessed at doses used in desvenlafaxine efficacy and safety studies57–62 from the recommended therapeutic dose of 50 mg/d to 400 mg/d (Table 3). Results from the desvenlafaxine CYP studies are described below and summarized in Table 4.

Effect of Desvenlafaxine
on CYP Substrate Drug Exposure
CYP 2D6. Coadministration of desvenlafaxine 100 mg/d with desipramine 50 mg resulted in increases in desipramine Cmax and AUC of 25% and 17%, respectively.20 The 90% CI for GM ratio for desipramine AUC (110%–125%) fell within the 80%–125% acceptance range for bioequivalence, indicating no interaction between these drugs. The ratios of GM AUC and Cmax values for the metabolite 2-hydroxydesipramine were 114% (90% CI, 110%–119%) and 110% (90% CI, 104%–116%), respectively.
Desvenlafaxine 100 mg/d was coadministered with desipramine in a second study that used a 4-period design for a comparison with paroxetine. In that study, Cmax and AUC increased by 30% and 36%, respectively, and the 90% CI for GM ratio for desipramine AUC (114%–163%) fell outside the 80%–125% bioequivalence range.21 The ratios of GM Cmax and AUC values for 2-hydroxydesipramine were 116% (90% CI, 100%–134%) and 100% (90% CI, 83%–121%), respectively. The 1.36-fold change in desipramine AUC indicates weak inhibition by desvenlafaxine 100 mg/d in that study. By comparison, coadministration with paroxetine 20 mg resulted in a > 5-fold increase in desipramine AUC (GM ratios [90% CI]: desipramine, 519% [433%–621%]; 2-hydroxydesipramine, 82% [71%–95%]).21
Coadministration with desvenlafaxine at the 400-mg/d dose resulted in desipramine Cmax and AUC increases of 52% and 90%, respectively; 2-hydroxydesipramine Cmax and AUC decreased by 18% (GM ratio: 82% [76%–88%]) and increased by 24% (GM ratio: 124% [119%–130%]), respectively. The 90% CIs for the GM ratio for desipramine AUC fell outside the bioequivalence range (190% [175%–208%]), and the 1.9-fold change in AUC indicates that desvenlafaxine at 8 times the recommended therapeutic dose is a weak inhibitor of CYP 2D6.
CYP 3A4. Coadministration of desvenlafaxine 50 mg/d with the CYP 3A4 substrate midazolam (4 mg) resulted in decreases in midazolam Cmax and AUC of 14% and 29%, respectively.22 The 90% CI for GM ratio for midazolam AUC (71% [65%–78%]) fell outside the bioequivalence range, but the 1.29-fold decrease indicated a weak interaction between the drugs. Coadministration with desvenlafaxine at the 400-mg/d dose had a similar effect on midazolam exposure: midazolam Cmax and AUC decreased by 16% and 31%, respectively. Again, the 90% CI for GM AUC ratio fell outside the bioequivalence range (69% [61%–78%]), with magnitude of change indicating a weak interaction between midazolam and the 400-mg/d desvenlafaxine dose. Exposure to the metabolite 1′-hydroxymidazolam was unchanged after coadministration with either desvenlafaxine dose (GM ratio for AUC with desvenlafaxine 50 mg/d: 93% [87%–98%], with desvenlafaxine 400 mg/d: 98% [85%–113%]).
In this example, the change in AUC demonstrates reduced exposure after coadministration of the drugs, which could indicate weak induction of CYP 3A4 by desvenlafaxine. However, a CYP 3A4 promoter gene assay was conducted to test this hypothesis, and results showed no evidence of CYP 3A4 induction by desvenlafaxine.63 Further, the induction of CYP 3A4 would be expected to increase exposure to the substrate’s metabolite; the reduction in 1′-hydroxymidazolam AUC is therefore not consistent with induction of CYP 3A4. At present, the mechanism underlying the small decrease in midazolam exposure after concomitant administration of desvenlafaxine is unknown.
CYP 2D6 and CYP 3A4. Results for coadministration of tamoxifen 40 mg with desvenlafaxine 100 mg/d indicated little potential for clinically significant DDI for a drug sequentially metabolized via first CYP 2D6 and then CYP 3A4. Coadministration with steady-state desvenlafaxine 100 mg resulted in changes in the ratios of adjusted geometric means for tamoxifen Cmax and AUC < 1% each, and 90% CIs for the AUC GM ratio fell within the bioequivalence range (GM ratio: 101% [97%–105%]).23 For the active metabolite endoxifen, the ratio of GMs for AUC was 88% (83%–94%; corrected for carry-over administration of tamoxifen alone).
Coadministration of aripiprazole 5 mg with steady-state desvenlafaxine 100 mg resulted in minimal change in aripiprazole exposure. Aripiprazole Cmax and AUC increased 1% and 6%, respectively. Coadministration increased dehydroaripiprazole Cmax and AUC by 6% and 5%, respectively. The GM ratio for aripiprazole AUC was 106%, and its 90% CIs (101%–111%) fell within the bioequivalence range.24 Little potential for clinically significant DDI with desvenlafaxine is observed for drugs metabolized by both the CYP 2D6 and CYP 3A4 pathways.
Effect of CYP 3A4
Inhibition on Desvenlafaxine Exposure
Coadministration with ketoconazole increased the Cmax and AUC of desvenlafaxine 400 mg by 8% and 43%, respectively.22 The 90% CI for the GM ratio for desvenlafaxine AUC fell outside the bioequivalence range, but the interaction between desvenlafaxine and the CYP 3A4 inhibitor was weak, based on the 1.43-fold increase in desvenlafaxine AUC.
UGT Isozymes
To determine the number of UGT isozymes that can catalyze glucuronidation of a new drug, and therefore the likelihood of DDI at the UGT pathway level, the new drug is incubated with cells expressing specific human UGTs, and drug and glucuronide metabolite levels are measured after incubation with a panel of different UGT isoforms. The presence of the glucuronide metabolite after incubation with a specific UGT isoform confirms that the isoform can mediate glucuronidation of the new drug. If preclinical studies indicate that a single UGT catalyzes glucuronidation of the new drug, pharmacokinetic studies like those described for CYP pathways would be used to confirm interactions at that site.49
Involvement of UGT isoforms in desvenlafaxine metabolism was assessed in preclinical studies. Conversion from desvenlafaxine to the glucuronide metabolite, O-desmethylvenlafaxine glucuronide, was found to be mediated by multiple isozymes.64 The presence of multiple glucuronidation pathways for desvenlafaxine indicates that the potential for a clinically significant DDI mediated by any single UGT isoform is low, and, hence, pharmacokinetic studies are not necessary.
P-Gp
The status of a drug as an inhibitor or substrate of P-gp can be assessed using in vitro methods such as a bidirectional transport assay. Per FDA guidance, a drug is a poor or non–P-gp substrate if the net efflux ratio comparing transport of the potential P-gp substrate drug across a cellular monolayer expressing the transporter in the basolateral to apical versus the apical to basolateral direction is less than 2.1,65 Inhibition of P-gp is assessed by measuring the transport of a known substrate drug across the monolayer in the presence of increasing concentrations of the potential inhibitor.1,65 A lack of P-gp inhibition can be concluded if no effect of increasing test-drug concentration on net flux ratio of the substrate is observed, or based on 50% inhibitory drug concentration (IC50) if inhibition is observed at high test-drug concentrations.1,65
Desvenlafaxine interaction with P-gp was studied in Caco-2 cell monolayers using the model P-gp substrate35 digoxin (5 μM) and the known P-gp inhibitor1,66 verapamil (100 μM) as a positive control.25 The efflux ratio for desvenlafaxine was < 2 (1.3–1.5) at all doses tested (5, 25, and 100 μM), indicating that desvenlafaxine is not a P-gp substrate.25 Further, desvenlafaxine was not an inhibitor of P-gp activity in the Caco-2 assay: minimal inhibition was observed at the highest desvenlafaxine concentration used in the inhibition studies (250 μM), and the IC50 could not be calculated, as inhibition at all desvenlafaxine concentrations was < 20%.25
CONCLUSIONS
In an illustration of the assessment of the potential for DDIs, a series of studies demonstrated a low potential for DDIs with desvenlafaxine through the CYP, UGT, and P-gp systems. In vivo pharmacokinetic studies showed either no signal or weak interaction based on FDA guidance with 2D6, 3A4, or the combination. Preclinical studies demonstrated that multiple UGT isozymes contribute to the phase II metabolism of desvenlafaxine, resulting in low risk of DDI via that pathway. Desvenlafaxine had clinically insignificant effects on the activity of P-gp in in vitro studies; on the basis of FDA guidance, there is no need to carry out in vivo studies when no in vitro signal is observed.1 The low potential for pharmacokinetically mediated DDI with desvenlafaxine is clinically important in light of the high rate of multiple medication use in patients who take antidepressant drugs.3,18 Consistent with the findings of the DDI studies reported here, no case studies describing DDIs involving patients taking desvenlafaxine have been published to date.
Patients prescribed antidepressant drugs represent a large percentage of the overall US population, making the potential for DDIs a public as well as individual concern. Antidepressants are among the most widely prescribed medications in the United States,67 and more than half of all antidepressant medications are prescribed by primary care physicians, who may be less familiar than specialists with DDI risks associated with antidepressant drugs.68–70 In a naturalistic study of 900 depressed patients prescribed venlafaxine, a total of 705 of 900 patients (78%) took at least 1 other drug in addition to the antidepressant18 (Table 1). Such high rates of concomitant medication use underscore the importance of educating prescribers about how the process outlined in the FDA guidance can inform them of (1) whether an interaction occurs and (2), if so, how to judge to what degree it is likely to be clinically meaningful.
While the studies reviewed here are essential for providing an assessment of the potential for clinically significant DDIs in the general population of patients prescribed a new antidepressant medication, there are specific patient populations and drugs for which the clinical significance of a DDI may be increased. Patients with a reduced capacity to metabolize a particular drug are at greater risk for clinically significant DDI, and a critical metabolic pathway can be compromised by renal disease, liver disease, or poor or intermediate CYP metabolizer status. Further, small pharmacokinetic interactions may be clinically significant for drugs with a narrow therapeutic range,17 as there is little separation between therapeutic and toxic doses of these drugs; a concomitant medication that minimally increases exposure can produce a clinically important DDI with a drug with a narrow therapeutic range. Clinicians should therefore assess DDI risk for each patient individually. In some cases, therapeutic drug monitoring can be an important tool in the practice of “personalized medicine,” to determine which drug or drug combinations are most effective and best tolerated in specific patients.71,72 If response to antidepressant treatment is insufficient or unexpected adverse effects occur, assessing plasma drug levels can indicate if the patient’s clearance is unsusually rapid or slow, and the possibility of reduced or extensive metabolism due to genotype or DDI should be considered. For such patients, an antidepressant drug with a low potential for DDI may be the best treatment option. With these caveats in mind when prescribing for individual MDD patients, clinicians should consider DDI analyses when assessing the potential for a drug to act as either a perpetrator or victim of a clinically meaningful, pharmacokinetically mediated DDI.
Drug names: alprazolam (Xanax, Niravam, and others), amlodipine (Norvasc and others), aripiprazole (Abilify), atomoxetine (Strattera), atorvastatin (Lipitor), buspirone (BuSpar and others), citalopram (Celexa and others), desipramine (Norpramin and others), desvenlafaxine (Pristiq), digoxin (Lanoxin and others), duloxetine (Cymbalta), eletriptan (relpaz), escitalopram (Lexapro and others), felodipine (Plendil and others), fluoxetine (Prozac and others), itraconazole (Sporanox, Onmel, and others), ketoconazole (Nizoral, and others), loperamide (Imodium), lovastatin (Altoprev and others), metoprolol (Toprol, Lopressor, and others), naproxen (Naprosyn and others), nelfinavir (Viracept), omeprazole (Prilosec and others), oxycodone (OxyContin, Roxicodone, and others), paroxetine (Paxil, Pexeva, and others), quetiapine (Seroquel), ritonavir (Norvir),sertraline (Zoloft and others), sildenafil (Viagra and Revatio), tacrolimus (Astagraf XL, Prograf), tamoxifen (Soltamox and others), triazolam (Halcion and others), venlafaxine (Effexor and others), verapamil (Verelan, Isoptin, and others), zolpidem (Ambien, Edluar, and others).
Author affiliations: University of Kansas School of Medicine, Wichita, Kansas (Drs Macaluso and Preskorn); Pfizer Inc, Collegeville, Pennsylvania (Dr Nichols); and Laureate Institute for Brain Research, Tulsa, Oklahoma
(Dr Preskorn).
Potential conflicts of interest: Dr Macaluso has participated in clinical trial research as principal investigator for AbbVie, Eisai, Envivo, Janssen, Naurex, and Pfizer; all clinical trial and study contracts were with and payments made to the Kansas University Medical Center Research Institute, a research institute affiliated with Kansas University School of Medicine–Wichita. Dr Nichols is currently employed by and receiving stock options from Pfizer. Dr Preskorn has served as a consultant to Abbott, AstraZeneca, Eisai, Johnson & Johnson, and Merck; has received grant/research funding from EnViVo, GlaxoSmithKline, NIMH, Pfizer, Stanley Medical Research Institute, and Sunovion; and has served on the scientific advisory board for AssureX.
Funding/support: The study from which data on file have been reported in Table 1 was sponsored by Wyeth Research, which was acquired by Pfizer in October 2009. Editorial and manuscript management was provided by Kathleen Dorries, PhD, Peloton Advantage, LLC, Parsippany, New Jersey, and was funded by Pfizer. Drs Macaluso and Preskorn did not receive remunerations of any kind for their work on this article.
Role of the sponsor: The manuscript was written by the authors without direct financial support from the sponsor. One author (A.I.N.) is an employee of the sponsor.
REFERENCES
1. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for Industry. Drug interaction studies: study design, data analysis, implications for dosing, and labeling recommendations. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf. Accessed May 20, 2013.
2. National Center for Health Statistics. Health, United States 2012 with Special Feature on Emergency Care. Hyattsville, MD: National Center for Health Statistics; 2013.
3. Silkey B, Preskorn SH, Golbeck A, et al. Complexity of medication use in the Veterans Affairs healthcare system, part 2: antidepressant use among younger and older outpatients. J Psychiatr Pract. 2005;11(1):16–26. doi:10.1097/00131746-200501000-00003 PubMed
4. Rush AJ, Kilner J, Fava M, et al. Clinically relevant findings from STAR*D. Psychiatr Ann. 2008;38(3):188–193. doi:10.3928/00485713-20080301-08
5. Warden D, Rush AJ, Trivedi MH, et al. The STAR*D Project results: a comprehensive review of findings. Curr Psychiatry Rep. 2007;9(6):449–459. doi:10.1007/s11920-007-0061-3 PubMed
6. Work Group on Major Depressive Disorder. Practice guideline for the treatment of patients with major depressive disorder. American Psychiatric Association. http://www.psychiatryonline.com/pracGuide/pracGuideTopic_7.aspx. Accessed February 1, 2012.
7. Rush AJ, Fava M, Wisniewski SR, et al; STAR*D Investigators Group. Sequenced treatment alternatives to relieve depression (STAR*D): rationale and design. Control Clin Trials. 2004;25(1):119–142. doi:10.1016/S0197-2456(03)00112-0 PubMed
8. Kessler RC, Berglund P, Demler O, et al; National Comorbidity Survey Replication. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095–3105. doi:10.1001/jama.289.23.3095 PubMed
9. Ettinger A, Reed M, Cramer J; Epilepsy Impact Project Group. Depression and comorbidity in community-based patients with epilepsy or asthma. Neurology. 2004;63(6):1008–1014. doi:10.1212/01.WNL.0000138430.11829.61 PubMed
10. Anderson RJ, Freedland KE, Clouse RE, et al. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care. 2001;24(6):1069–1078. doi:10.2337/diacare.24.6.1069 PubMed
11. Eaton WW. Epidemiologic evidence on the comorbidity of depression and diabetes. J Psychosom Res. 2002;53(4):903–906. doi:10.1016/S0022-3999(02)00302-1 PubMed
12. Halaris A. Comorbidity between depression and cardiovascular disease. Int Angiol. 2009;28(2):92–99. PubMed
13. González HM, Tarraf W. Comorbid cardiovascular disease and major depression among ethnic and racial groups in the United States. Int Psychogeriatr. 2013;25(5):833–841. doi:10.1017/S1041610212002062 PubMed
14. Kanner AM, Schachter SC, Barry JJ, et al. Depression and epilepsy: epidemiologic and neurobiologic perspectives that may explain their high comorbid occurrence. Epilepsy Behav. 2012;24(2):156–168. doi:10.1016/j.yebeh.2012.01.007 PubMed
15. Bair MJ, Robinson RL, Katon W, et al. Depression and pain comorbidity: a literature review. Arch Intern Med. 2003;163(20):2433–2445. doi:10.1001/archinte.163.20.2433 PubMed
16. Ligthart L, Gerrits MM, Boomsma DI, et al. Anxiety and depression are associated with migraine and pain in general: an investigation of the interrelationships. J Pain. 2013;14(4):363–370. doi:10.1016/j.jpain.2012.12.006 PubMed
17. Preskorn SH, Flockhart D. 2010 guide to psychiatric drug interactions. Prim Psychiatry. 2009;16(12):45–74.
18. Preskorn SH, Kane CP, Lobello K, et al. Cytochrome P450 2D6 phenoconversion is common in patients being treated for depression: implications for personalized medicine. J Clin Psychiatry. 2013;74(6):614–621. doi:10.4088/JCP.12m07807 PubMed
19. P450 Drug Interaction Table. Indiana University, Department of Medicine, Division of Clinical Pharmacology. http://medicine.iupui.edu/clinpharm/ddis/table.aspx. Updated July 12, 2013. Accessed January 25, 2012.
20. Nichols AI, Abell M, Chen Y, et al. Effects of desvenlafaxine on the pharmacokinetics of desipramine in healthy adults. Int Clin Psychopharmacol. 2013;28(2):99–105. doi:10.1097/YIC.0b013e32835c1f49 PubMed
21. Nichols AI, Fatato P, Shenouda M, et al. The effects of desvenlafaxine and paroxetine on the pharmacokinetics of the cytochrome P450 2D6 substrate desipramine in healthy adults. J Clin Pharmacol. 2009;49(2):219–228. doi:10.1177/0091270008326716 PubMed
22. Nichols A, Liang Y, Matschke K, et al. An evaluation of the potential of cytochrome P450 3A4-mediated drug-drug interactions with desvenlafaxine use. J Bioequiv Availab. 2013;5(1):53–59. doi:10.4172/jbb.1000134
23. Nichols AI, Lubaczewski S, Liang Y, et al. Open-label, 2-period sequential drug interaction study to evaluate the effect of a 100-mg dose of desvenlafaxine on the pharmacokinetics of tamoxifen when coadministered in healthy postmenopausal female subjects. Int J Clin Pharmacol Ther. 2014;52(10):830–841. doi:10.5414/CP201958 PubMed
24. Nichols AI, Lubaczewski S, Liang Y, et al. An open-label evaluation of the effect of coadministering desvenlafaxine 100 mg on the pharmacokinetics of aripiprazole in healthy subjects. J Bioequiv Availab. 2013;5(7):253–259.
25. Oganesian A, Shilling AD, Young-Sciame R, et al. Desvenlafaxine and venlafaxine exert minimal in vitro inhibition of human cytochrome P450 and P-glycoprotein activities. Psychopharmacol Bull. 2009;42(2):47–63. PubMed
26. Ingelman-Sundberg M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol Sci. 2004;25(4):193–200. doi:10.1016/j.tips.2004.02.007 PubMed
27. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464–494. doi:10.1176/appi.psy.46.5.464 PubMed
28. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34(1–2):83–448. doi:10.1081/DMR-120001392 PubMed
29. Takano M, Yumoto R, Murakami T. Expression and function of efflux drug transporters in the intestine. Pharmacol Ther. 2006;109(1–2):137–161. doi:10.1016/j.pharmthera.2005.06.005 PubMed
30. Glaeser H. Importance of P-glycoprotein for drug-drug interactions. Handb Exp Pharmacol. 2011;201(201):285–297. doi:10.1007/978-3-642-14541-4_7 PubMed
31. Holtzman CW, Wiggins BS, Spinler SA. Role of P-glycoprotein in statin drug interactions. Pharmacotherapy. 2006;26(11):1601–1607. doi:10.1592/phco.26.11.1601 PubMed
32. Huisman MT, Smit JW, Schinkel AH. Significance of P-glycoprotein for the pharmacology and clinical use of HIV protease inhibitors. AIDS. 2000;14(3):237–242. doi:10.1097/00002030-200002180-00005 PubMed
33. Kim WY, Benet LZ. P-glycoprotein (P-gp/MDR1)-mediated efflux of sex-steroid hormones and modulation of P-gp expression in vitro. Pharm Res. 2004;21(7):1284–1293. doi:10.1023/B:PHAM.0000033017.52484.81 PubMed
34. Yu DK. The contribution of P-glycoprotein to pharmacokinetic drug-drug interactions. J Clin Pharmacol. 1999;39(12):1203–1211. doi:10.1177/00912709922012006 PubMed
35. Akamine Y, Yasui-Furukori N, Ieiri I, et al. Psychotropic drug-drug interactions involving P-glycoprotein. CNS Drugs. 2012;26(11):959–973. doi:10.1007/s40263-012-0008-z PubMed
36. Stearns V, Johnson MD, Rae JM, et al. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst. 2003;95(23):1758–1764. doi:10.1093/jnci/djg108 PubMed
37. Jin Y, Desta Z, Stearns V, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005;97(1):30–39. doi:10.1093/jnci/dji005 PubMed
38. Poulsen L, Brøsen K, Arendt-Nielsen L, et al. Codeine and morphine in extensive and poor metabolizers of sparteine: pharmacokinetics, analgesic effect and side effects. Eur J Clin Pharmacol. 1996;51(3–4):289–295. doi:10.1007/s002280050200 PubMed
39. Lobello KW, Preskorn SH, Guico-Pabia CJ, et al. Cytochrome P450 2D6 phenotype predicts antidepressant efficacy of venlafaxine: a secondary analysis of 4 studies in major depressive disorder. J Clin Psychiatry. 2010;71(11):1482–1487. doi:10.4088/JCP.08m04773blu PubMed
40. Rau T, Wohlleben G, Wuttke H, et al. CYP2D6 genotype: impact on adverse effects and nonresponse during treatment with antidepressants: a pilot study. Clin Pharmacol Ther. 2004;75(5):386–393. doi:10.1016/j.clpt.2003.12.015 PubMed
41. Goetz MP, Knox SK, Suman VJ, et al. The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Res Treat. 2007;101(1):113–121. doi:10.1007/s10549-006-9428-0 PubMed
42. de Leon J, Susce MT, Pan RM, et al. The CYP2D6 poor metabolizer phenotype may be associated with risperidone adverse drug reactions and discontinuation. J Clin Psychiatry. 2005;66(1):15–27. doi:10.4088/JCP.v66n0103 PubMed
43. Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron. 2008;57(2):203–209. doi:10.1016/j.neuron.2007.11.017 PubMed
44. Kelly CM, Juurlink DN, Gomes T, et al. Selective serotonin reuptake inhibitors and breast cancer mortality in women receiving tamoxifen: a population based cohort study. BMJ. 2010;340:c693. doi:10.1136/bmj.c693 PubMed
45. Borges S, Desta Z, Li L, et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin Pharmacol Ther. 2006;80(1):61–74. doi:10.1016/j.clpt.2006.03.013 PubMed
46. Desta Z, Ward BA, Soukhova NV, et al. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther. 2004;310(3):1062–1075. doi:10.1124/jpet.104.065607 PubMed
47. Lim YC, Li L, Desta Z, et al. Endoxifen, a secondary metabolite of tamoxifen, and 4-OH-tamoxifen induce similar changes in global gene expression patterns in MCF-7 breast cancer cells. J Pharmacol Exp Ther. 2006;318(2):503–512. doi:10.1124/jpet.105.100511 PubMed
48. Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J. 2003;3(3):136–158. doi:10.1038/sj.tpj.6500171 PubMed
49. Kiang TK, Ensom MH, Chang TK. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther. 2005;106(1):97–132. doi:10.1016/j.pharmthera.2004.10.013 PubMed
50. Harper TW, Brassil PJ. Reaction phenotyping: current industry efforts to identify enzymes responsible for metabolizing drug candidates. AAPS J. 2008;10(1):200–207. doi:10.1208/s12248-008-9019-6 PubMed
51. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals, Inc, a subsidiary of Pfizer Inc; 2013.
52. Preskorn SH, Nichols AI, Paul J, et al. Effect of desvenlafaxine on the cytochrome P450 2D6 enzyme system. J Psychiatr Pract. 2008;14(6):368–378. doi:10.1097/01.pra.0000341891.43501.6b PubMed
53. Ereshefsky L, Jhee S, Grothe D. Antidepressant drug-drug interaction profile update. Drugs R D. 2005;6(6):323–336. doi:10.2165/00126839-200506060-00002 PubMed
54. Food and Drug Administration. Drug development and drug interactions: table of substrates, inhibitors and inducers. FDA web site. http://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm. Accessed September 4, 2014.
55. Winans E. Aripiprazole.Am J Health Syst Pharm. 2003;60(23):2437–2445. PubMed
56. Huang SM, Temple R, Throckmorton DC, et al. Drug interaction studies: study design, data analysis, and implications for dosing and labeling. Clin Pharmacol Ther. 2007;81(2):298–304. doi:10.1038/sj.clpt.6100054 PubMed
57. DeMartinis NA, Yeung PP, Entsuah R, et al. A double-blind, placebo-controlled study of the efficacy and safety of desvenlafaxine succinate in the treatment of major depressive disorder. J Clin Psychiatry. 2007;68(5):677–688. doi:10.4088/JCP.v68n0504 PubMed
58. Septien-Velez L, Pitrosky B, Padmanabhan SK, et al. A randomized, double-blind, placebo-controlled trial of desvenlafaxine succinate in the treatment of major depressive disorder. Int Clin Psychopharmacol. 2007;22(6):338–347. doi:10.1097/YIC.0b013e3281e2c84b PubMed
59. Liebowitz MR, Manley AL, Padmanabhan SK, et al. Efficacy, safety, and tolerability of desvenlafaxine 50 mg/day and 100 mg/day in outpatients with major depressive disorder. Curr Med Res Opin. 2008;24(7):1877–1890. doi:10.1185/03007990802161923 PubMed
60. Boyer P, Montgomery S, Lepola U, et al. Efficacy, safety, and tolerability of fixed-dose desvenlafaxine 50 and 100 mg/day for major depressive disorder in a placebo-controlled trial. Int Clin Psychopharmacol. 2008;23(5):243–253. doi:10.1097/YIC.0b013e32830cebed PubMed
61. Iwata N, Tourian KA, Hwang E, et al; for the Study 3359 Investigators. Efficacy and safety of desvenlafaxine 25 and 50 mg/day in a randomized, placebo-controlled study of depressed outpatients. J Psychiatr Pract. 2013;19(1):5–14. doi:10.1097/01.pra.0000426323.59698.64 PubMed
62. Dunlop BW, Reddy S, Yang L, et al. Symptomatic and functional improvement in employed depressed patients: a double-blind clinical trial of desvenlafaxine versus placebo. J Clin Psychopharmacol. 2011;31(5):569–576. doi:10.1097/JCP.0b013e31822c0a68 PubMed
63. DeMaio W, Kane CP, Nichols AI, et al. Metabolism studies of desvenlafaxine. J Bioequiv Availab. 2011;3(7):151–160. doi:10.4172/jbb.1000076
64. Nichols AI, Tourian KA, Tse SY, et al. Desvenlafaxine for major depressive disorder: incremental clinical benefits from a second-generation serotonin-norepinephrine reuptake inhibitor. Expert Opin Drug Metab Toxicol. 2010;6(12):1565–1574. doi:10.1517/17425255.2010.535810 PubMed
65. Grandvuinet AS, Vestergaard HT, Rapin N, et al. Intestinal transporters for endogenic and pharmaceutical organic anions: the challenges of deriving in-vitro kinetic parameters for the prediction of clinically relevant drug-drug interactions. J Pharm Pharmacol. 2012;64(11):1523–1548. doi:10.1111/j.2042-7158.2012.01505.x PubMed
66. Linnet K, Ejsing TB. A review on the impact of P-glycoprotein on the penetration of drugs into the brain: focus on psychotropic drugs. Eur Neuropsychopharmacol. 2008;18(3):157–169. doi:10.1016/j.euroneuro.2007.06.003 PubMed
67. Gill JM, Klinkman MS, Chen YX. Antidepressant medication use for primary care patients with and without medical comorbidities: a national electronic health record (EHR) network study. J Am Board Fam Med. 2010;23(4):499–508. doi:10.3122/jabfm.2010.04.090299 PubMed
68. Cascade E, Kalali AH, Halbreich U. Antidepressant prescribing by specialty and treatment of premenstrual dysphoric disorder. Psychiatry (Edgmont). 2008;5(12):14–15. PubMed
69. Mark TL, Levit KR, Buck JA. Datapoints: psychotropic drug prescriptions by medical specialty. Psychiatr Serv. 2009;60(9):1167. doi:10.1176/appi.ps.60.9.1167 PubMed
70. Milea D, Verpillat P, Guelfucci F, et al. Prescription patterns of antidepressants: findings from a US claims database. Curr Med Res Opin. 2010;26(6):1343–1353. doi:10.1185/03007991003772096 PubMed
71. Hiemke C, Baumann P, Bergemann N, et al. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry. 2011;44(6):195–235. doi:10.1055/s-0031-1286287
72. Preskorn SH. Therapeutic Drug Monitoring (TDM) in psychiatry (part 1): why studies attempting to correlate drug concentration and antidepressant response don’t work. J Psychiatr Pract. 2014;20(2):133–137. doi:10.1097/01.pra.0000445247.54048.68 PubMed
Enjoy this premium PDF as part of your membership benefits!


