A Novel Mutation of the Arylsulfatase A Gene in Late-Onset Metachromatic Leukodystrophy
To the Editor: Metachromatic leukodystrophy (MLD) is a rare lysosomal storage disorder that is caused by a deficiency of the enzyme arylsulfatase A (ARSA). Toxic accumulation of sulfatides in myelin-producing cells of the central nervous system and peripheral nervous system (PNS) results in progressive demyelination.1 The 3 major forms of MLD are late infantile-, juvenile-, and adult-onset MLD. Here, we report a novel mutation of the ARSA gene in a patient with late-onset MLD, reminiscent of rapid progressive frontotemporal dementia without signs of peripheral neuropathy.
Case report. Ms A, a 37-year-old Ukrainian woman, was the single child of nonconsanguineous parents. No developmental abnormalities were reported. At the age of 32 years, the first signs of personality change were noticed. Two years later, slight memory deficits, uncontrolled eating, inadequate social behavior, and sexual disinhibition occurred, and the patient was dismissed by her employer. Five years after the onset of first symptoms, Ms A was admitted to a nursing home due to progressive apathy, loss of spontaneous speech, neglect of personal hygiene, and neglect of her 1-year-old son.
Neuropsychological testing showed frontal signs in addition to severely impaired cognitive function, including deficits of visuospatial orientation and memory. Cranial magnetic resonance images revealed moderate global brain atrophy. T2-weighted sequences showed diffuse bilateral and confluent hyperintensities with frontal and temporoparietal accentuation (Figure 1A). The scans, including diffusion-weighted magnetic resonance imaging, demonstrated demyelination and volume reduction of the corpus callosum. These lesions were reminiscent of and consistent with the pattern observed in MLD. In addition, we found involvement of subcortical U-fibers and striatal atrophy, a finding that is not commonly observed early in MLD and rather seen in late disease stages. The striatal atrophy is rarely seen in MLD and might contribute to the predominant frontal syndrome, since the striatum is a principal structure in frontal-subcortical anatomic circuits that mediate cognitive and emotional functions. Proton spectroscopy of corpus callosum and periventricular regions revealed increased intralesional choline and decreased N-acetyl aspartate signals, compatible with the diagnosis of MLD. The spectroscopic data are indicative of significant neuronal loss in addition to demyelination. The diagnosis of MLD was confirmed by increased urinary sulfatide excretion and moderately reduced ARSA activity in peripheral blood leukocytes (10.93 nmol/h/mg, compared to the normal range of 31-151 nmol/h/mg). Neurologic examination results and peripheral nerve conduction velocities were normal. Sequence analysis of genomic leukocyte DNA revealed compound heterozygosity for the novel Phe144Leu mutation and the known mutation Ile179Ser.
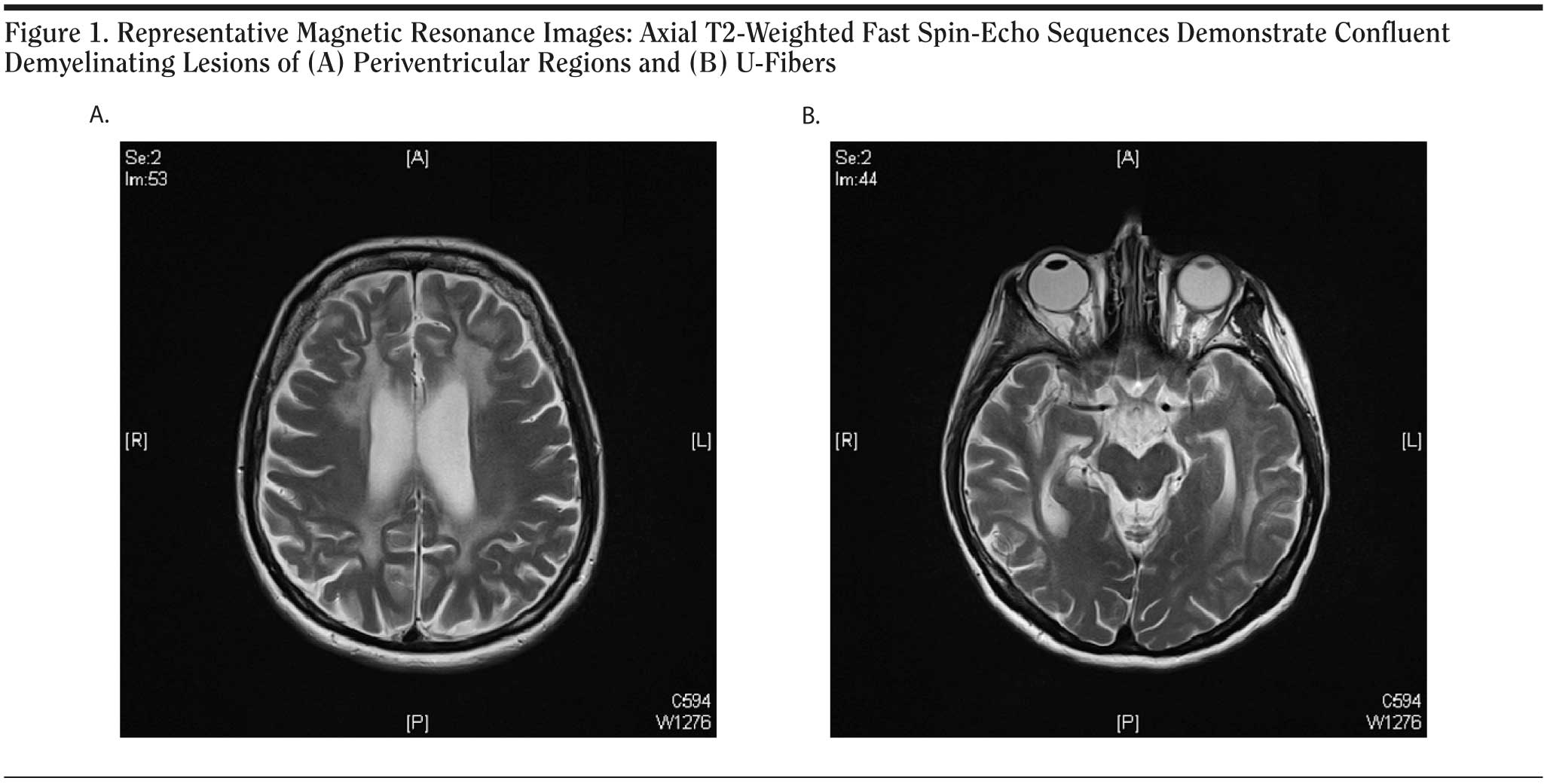
Click figure to enlarge
Reduced nerve conduction velocities are commonly present early in the disease course of MLD, and few cases without electrophysiologic PNS abnormalities have been reported (eg, Marcao et al2). The presented case shows an unusual combination of rapid cognitive decline in the absence of PNS involvement. Adult-onset MLD progresses slowly, sometimes even over decades. The rapid disease progress with severe frontal lobe signs initially prompted us to suspect frontotemporal dementia. While psychotic symptoms are common in adult-onset MLD, a frontotemporal dementia-like disease course has rarely been described.3
In summary, we report a novel mutation of the ARSA gene that is associated with rapid disease progress in the absence of peripheral neuropathy. This case indicates that MLD needs to be considered in the differential diagnosis of frontal-subcortical dementia syndrome.
References
1. Gieselmann V. Metachromatic leukodystrophy: genetics, pathogenesis and therapeutic options. Acta Paediatr Suppl. 2008;97(s457):15-21. PubMed doi:10.1111/j.1651-2227.2008.00648.x
2. Marcao AM, Wiest R, Schindler K, et al. Adult onset metachromatic leukodystrophy without electroclinical peripheral nervous system involvement: a new mutation in the ARSA gene. Arch Neurol. 2005;62(2):309-313. PubMed doi:10.1001/archneur.62.2.309
3. Kozian R, Sieber N, Thiergart S. Frontotemporale Demenz bei metachromatischer Leukodystrophie [Frontotemporal dementia in metachromatic leukodystrophy]. Fortschr Neurol Psychiatr. 2007;75(9):549-551. PubMed doi:10.1055/s-2007-980065
Author affiliations: Memory Clinic, Department of Psychiatry and Psychotherapy (Drs Schneider and Degner and Ms Hirschel); Department of Psychiatry and Psychotherapy (Drs Hasan, Falkai, and Wobrock); and Department of Pediatrics (Drs Gפrtner and Steinfeld), University Medicine, University of Goettingen; and Center for Human Genetics, Freiburg (Drs Wilhelm and Kohlhase), Germany. Financial disclosure: None reported. Funding/support: None reported.
doi:10.4088/JCP.09l05010
© Copyright 2009 Physicians Postgraduate Press, Inc.





{kind=link}