Inflammation is an important mediator of pathophysiology in bipolar disorder. The omega-3 (n-3) and omega-6 (n-6) polyunsaturated fatty acid (PUFA) metabolic pathways participate in several inflammatory processes and have been linked through epidemiologic and clinical studies to bipolar disorder and its response to treatment. We review the proposed role of PUFA metabolism in neuroinflammation, modulation of brain PUFA metabolism by antimanic medications in rodent models, and anti-inflammatory pharmacotherapy in bipolar disorder and in major depressive disorder (MDD). Although the convergence of findings between preclinical and postmortem clinical data is compelling, we investigate why human trials of PUFA as treatment are mixed. We view the biomarker and treatment study findings in light of the evidence for the hypothesis that arachidonic acid hypermetabolism contributes to bipolar disorder pathophysiology and propose that a combined high n-3 plus low n-6 diet should be tested as an adjunct to current medication in future trials.
See article by Saunders et al
Reconsidering Dietary Polyunsaturated Fatty Acids
in Bipolar Disorder:
A Translational Picture
ABSTRACT
Inflammation is an important mediator of pathophysiology in bipolar disorder. The omega-3 (n-3) and omega-6 (n-6) polyunsaturated fatty acid (PUFA) metabolic pathways participate in several inflammatory processes and have been linked through epidemiologic and clinical studies to bipolar disorder and its response to treatment. We review the proposed role of PUFA metabolism in neuroinflammation, modulation of brain PUFA metabolism by antimanic medications in rodent models, and anti-inflammatory pharmacotherapy in bipolar disorder and in major depressive disorder (MDD). Although the convergence of findings between preclinical and postmortem clinical data is compelling, we investigate why human trials of PUFA as treatment are mixed. We view the biomarker and treatment study findings in light of the evidence for the hypothesis that arachidonic acid hypermetabolism contributes to bipolar disorder pathophysiology and propose that a combined high n-3 plus low n-6 diet should be tested as an adjunct to current medication in future trials.
J Clin Psychiatry 2016;77(10):e1342–e1347
dx.doi.org/10.4088/JCP.15com10431
© Copyright 2016 Physicians Postgraduate Press, Inc.
aDepartment of Psychiatry, Penn State College of Medicine and Penn State Milton S. Hershey Medical Center, Hershey, Pennsylvania
bDepartment of Psychiatry and Depression Center, University of Michigan, Ann Arbor
cSection on Nutritional Neurosciences, Laboratory of Membrane Biochemistry and Biophysics, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, Maryland
dDepartment of Psychiatry, University of Illinois, Chicago
eOffice of Scientific Director, National Institute on Aging, National Institutes of Health, Bethesda, Maryland
*Corresponding author: Erika F. H. Saunders, MD, Department of Psychiatry, Penn State Milton S. Hershey Medical Center, Penn State College of Medicine, 500 University Dr, PO Box 850, Mail Code: HO73, Hershey, PA 17033-0850 ([email protected]).
Bipolar disorder affects 1%–4.4% of the population and has an episodic, recurrent course that causes significant disability and has a complex, incompletely understood etiology.1 Effective pharmacotherapies for acute episodes and prevention of relapse include lithium salts, certain antiepileptic agents (eg, valproic acid, carbamazepine, and lamotrigine), and antipsychotic agents.2
Although mood-stabilizing medications come from several pharmaceutical classes with differing primary mechanisms of action, downregulation of brain metabolism of arachidonic acid (AA, 20:4n-6), a long-chain omega-6 (n-6) polyunsaturated fatty acid (PUFA), has been suggested from preclinical studies as one common effect of mood-stabilizing medications.3–5 Linoleic acid (18:2n-6) is the dietary-essential shorter-chain n-6 PUFA precursor of AA, which is also consumed in the diet. Alterations of turnover of PUFAs in membrane lipids, including AA, and resultant alterations in cell-signaling pathways in brain cell membranes have long been hypothesized to be central to perturbations of neurotransmitter systems in mood disorders.6,7
Both studies of biological concentrations of PUFAs, circulating in the plasma or incorporated into red blood cell membranes, and treatment trials using omega-3 (n-3) PUFAs as dietary supplements have had mixed results in bipolar disorder. While heterogeneity in methods and treatment intervention are confounding factors, one additional reason may be that trials did not also involve alterations in dietary n-6 as well as n-3 PUFA intake. Preclinical studies suggest that neurotransmission and other brain functions depend on a balance between n-6 and n-3 PUFAs and their downstream metabolites, such as proinflammatory prostaglandins, lipoxins, and thromboxanes and anti-inflammatory resolvins and neuroprotectins, respectively. We present the biological underpinnings of PUFA-related interventions in a physiological, biochemical, and molecular context. To do this, we drew upon literature from preclinical animal work, postmortem brain studies, and parallel investigations in major depressive disorder (MDD).
We will review evidence for the proposed role of PUFA metabolism in neuroinflammation, modulation of brain PUFA metabolism by antimanic medications in rodent models, and anti-inflammatory pharmacotherapy in bipolar disorder and in MDD. On the basis of the reviewed evidence, we propose that dietary manipulation combining high n-3 PUFA with low n-6 PUFA should be tested as an adjunct to traditional mood-stabilizing medications in future clinical trials, rather than using a simple high n-3 PUFA–containing diet.
PUFAs and Neuroinflammation
The long-chain PUFAs AA and docosahexaenoic acid (DHA, 22:6n-3) comprise over 90% of PUFAs in the mammalian brain.8 Arachidonic acid and DHA can be either derived from the diet or synthesized in the liver from their respective nutritionally essential shorter-chain PUFAs, linoleic acid and α-linolenic acid (18:3n-3). AA, DHA, and their metabolites function as intracellular second messengers and as modulators of neuroinflammation, neurotransmission, gene transcription, and other important brain process.6 Proinflammatory cytokines can stimulate release of AA from membrane phospholipids, which then is available for metabolism by cyclooxygenase (COX)-1 or COX-2 to proinflammatory prostaglandins (eg, prostaglandin E2), by lipoxygenases to cytotoxic leukotrienes, or by cytochrome p450 epoxygenases to cytoprotective epoxyeicosatrienoic acids with the AA metabolic cascade (Figure 1).9 Prostaglandin E2 affects sleep, and may mediate pain pathways and interleukin-1–induced “sickness behavior,” which includes suppressed appetite, social withdrawal, psychomotor retardation, and poor concentration, symptoms that overlap with depression.10–12 Prostaglandins also regulate the hypothalamic pituitary adrenal axis by inducing corticotropin-releasing hormone release.13,14 Acetylsalicylic acid, a COX-1 inhibitor and COX-2 inhibitor and acetylator,15 reduces the cortisol response,16 and in a rat neuroinflammation model, chronic low-equivalent dose aspirin reduced brain levels of prostaglandin E2 and 8-isoprostane.17
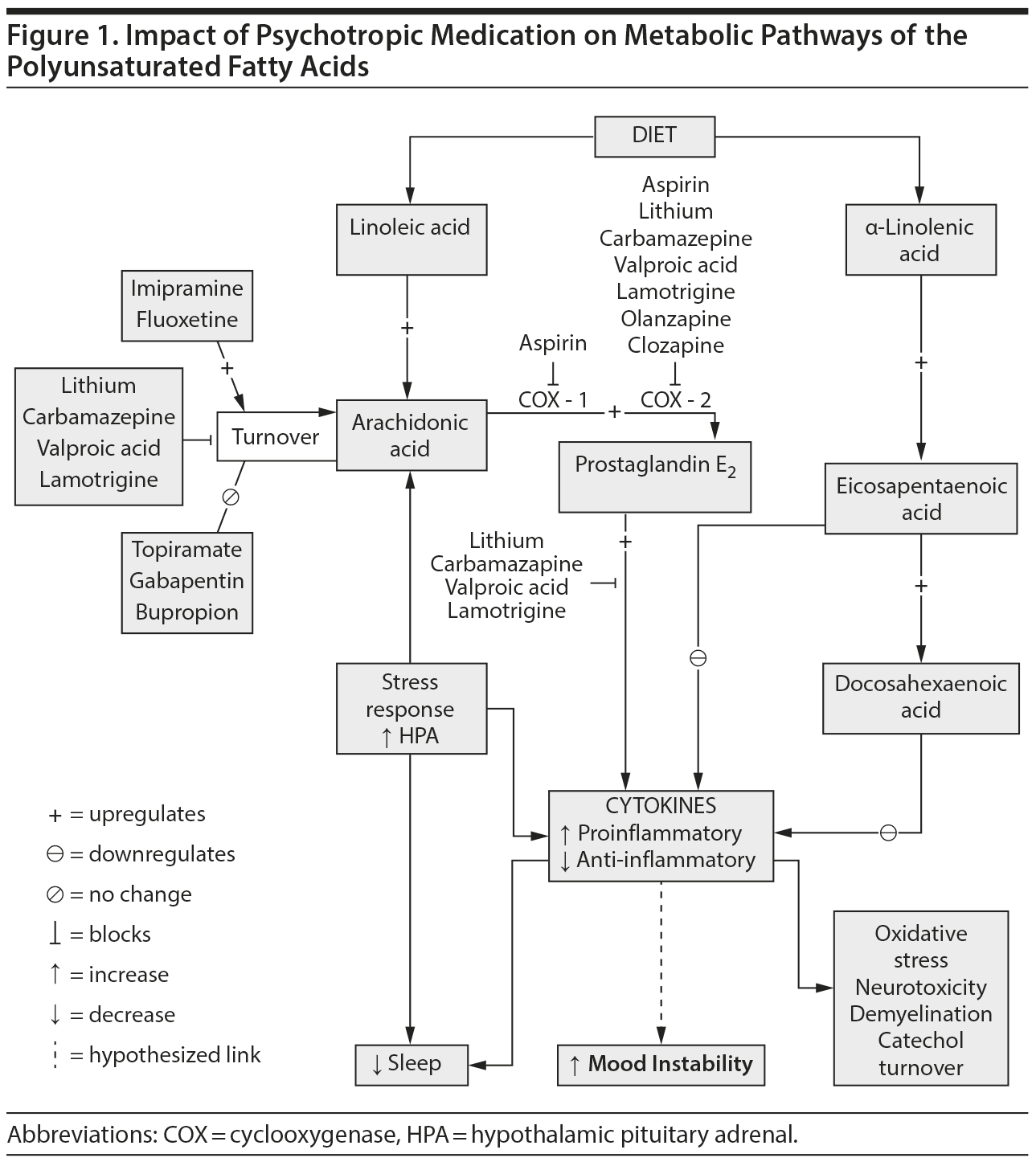
Arachidonic acid is hydrolyzed from membrane phospholipids by cytosolic or secretory phospholipase A2 (PLA2). While early genetic linkage studies18,19 of the chromosomal region coding for secretory PLA2 were promising, subsequent studies20–23 failed to find significant association between PLA2 genes and bipolar disorder. Serum PLA2 levels were reported to be elevated in schizophrenia, bipolar disorder, MDD, posttraumatic stress disorder, and substance abuse.24 While a subsequent study25 showed no difference between bipolar disorder and control in enzyme activity of PLA2, calcium-independent PLA2 was elevated in patients with bipolar disorder and a history of psychosis compared to those without psychosis. A recent study,26 however, showed lower enzyme activity of 3 PLA2 species in platelet membranes of drug-naive bipolar disorder subjects compared to controls. Increased AA metabolism in bipolar disorder has been suggested by postmortem brain studies (eg, Kim et al27). Compared to controls, the frontal cortex of bipolar disorder patients had increased expression of some AA metabolism enzymes, including AA-selective cytosolic PLA2-IVA, secretory PLA2-IIA, COX-2, and membrane prostaglandin E synthase (PGES), while expression of others (COX-1 and cytosolic PGES) was reduced and of others (calcium-independent PLA2-VIA; 5-, 12-, and 15-lipoxigenase; thromboxane synthase; and cytochrome p450 epoxygenase) was unchanged,27 suggesting an increase in activity in the AA cascade.18,21,22 In this regard, a rat model of excitotoxicity using chronic administration of N-methyl-d-aspartate, a glutamate receptor agonist, showed increased cytosolic PLA2 and increased AA signaling in the frontal cortex.28,29
Data from postmortem human brain and animal models are consistent with the proposition that neuroinflammatory processes could contribute to disease progression in bipolar disorder. Upregulated markers of the proinflammatory AA cascade, which activate pathways leading to cell dysfunction and death, have been reported in postmortem human brain.27,30,31 Excitotoxic and proapoptotic factors were elevated and antiapoptotic and synaptic markers were decreased in the frontal cortex of those with bipolar disorder compared to controls.30,32 Studies33–40 of peripheral PUFA markers in bipolar disorder show converging findings that abnormalities in the n-6 and n-3 metabolism pathways are present in bipolar disorder.
Neuroinflammation associated with excitotoxicity and apoptosis in bipolar disorder may promote future episodes and disease worsening. Although bipolar disorder is an episodic illness, progressive changes in cognitive function and brain structure occur and correlate with severity and chronicity of illness. Neuroinflammation and excitotoxicity, leading to neuronal apoptosis and synaptic loss, have been hypothesized to underlie progression of bipolar disorder (reviewed in Berk et al41). Subtle cognitive changes are reported during and between episodes and are related to number and amount of time in manic episodes.42–44 Peripheral markers of inflammation are elevated in bipolar disorder.45,46 Neuroimaging studies47–49 have also reported brain atrophy and gray matter deficits in emotion regulation circuits linked to length of illness. In vivo imaging studies using functional magnetic resonance imaging have shown hemodynamic changes in the right amygdala and ventromedial prefrontal cortex and left hippocampus in euthymic bipolar disorder in response to an emotion task, and activation correlated with gene expression in inflammatory pathways.50 In another study,51 peripheral markers of inflammation in the kynurenine pathway were correlated with hippocampal volume. Additionally, a study52 using positron emission tomography has shown elevated markers of microglial activation in bipolar disorder in the hippocampus.
Upregulation of the AA cascade in bipolar disorder also could modulate signal transduction and interfere with synaptic function53 to promote worsening of illness and cognitive changes associated with duration of illness. Changes in the balance between the n-3 and n-6 PUFAs and their bioactive lipid autacoid derivatives (Figure 1) most likely influence the inflammatory response.36
Antimanic Medications Alter AA Metabolism
If upregulation of the AA cascade and subsequent neuroinflammation are associated with the pathophysiology and progression of bipolar disorder, effective treatments for bipolar disorder might act by downregulating the brain AA cascade.3–5 A study supporting this proposition found that chronic administration to rats of a therapeutically relevant dose of lithium reduced AA turnover in brain phospholipids, expression of important AA-metabolizing enzymes, and generation of prostaglandin E2,5 while not affecting DHA and/or palmitic acid (16:0) turnover.54 Other antiepileptic drugs with clinically proven antimanic efficacy (carbamazepine, valproate, lamotrigine) also downregulated the rat brain AA metabolic cascade,55–57 while topiramate or gabapentin, antiepileptic medications that have failed phase 3 trials, did not.4,58–60 Synthesizing these data has led to the hypothesis that therapeutic downregulation of the AA cascade can be tied specifically to effective treatment of bipolar disorder. Intervention could involve drugs (as discussed in this paragraph) as well and a change in the PUFA content of the diet (see The Way Forward).
Pharmacotherapy in Bipolar Disorder:
Anti-Inflammatory and n-3 PUFA Agents
In addition to the proven effective mood stabilizers, other pharmaceutical agents that interfere with brain AA metabolism would be of interest to investigate clinically in bipolar disorder. They might include acetylsalicylic acid and other nonsteroidal anti-inflammatory drugs (NSAIDs) (eg, nonselective COX inhibitors) or selective COX-2 or COX-1 inhibitors. Stolk et al61 retrospectively used a Netherlands database to investigate effects of some of these agents in subjects on lithium. Long-term low-dose aspirin was associated with reduced risk for worse outcomes, while short-term use of a nonselective NSAID, or of more than 1 inhibitor, increased risk. Nery et al62 found that celecoxib, a selective COX-2 inhibitor for treatment of depression or mixed episode in bipolar disorder, reduced severity of depression at 1 week, but did not have a sustained effect in a 6-week, double-blind, randomized controlled trial as an adjunct to usual treatment. The effect of anti-inflammatory treatments on mood outcome in bipolar disorder needs further consideration.
Studies in rodents report differing actions of antidepressants compared with mood-stabilizer medication on the brain AA and cytokine-based inflammatory systems. In unanesthetized rats, chronic imipramine and fluoxetine (a selective serotonin reuptake inhibitor [SSRI]), antidepressants that can increase risk for switching from depression to mania in bipolar disorder patients,63 upregulated brain AA turnover and metabolism (opposite to direction of changes with mood stabilizers), but bupropion, an antidepressant causing lower switch rates, did not.64,65 These comparisons suggest that increased brain AA metabolism may be associated with the manic phase of bipolar disorder and that stimulating AA metabolism may be associated with a switch of depression to mania with certain antidepressants. Indeed, coadministration of lithium, which depresses AA metabolism in rats, is recommended when fluoxetine is administered66; it might dampen the untoward AA upregulation of the SSRI. In mice, SSRIs increased inflammatory markers tumor necrosis factor-α, interferon (IFN)-γ, and p11 in the frontal cortex.67 Because of the complex interactions between AA metabolism and inflammation, the clinical implications of these findings remain to be elucidated. Interestingly, a study of n-3 to prevent IFN-α–induced depression in 162 patients treated for hepatitis C showed lower rates of IFN-α–induced depression in EPA- but not DHA-treated patients, and both n-3 treatments delayed the onset of depression.68
Studies of treatment of bipolar disorder with supplementation of n-3 preparations, either EPA or DHA or both, have been mixed, and several recent reviews33,69 have discussed these studies in detail. Briefly, open-label and nonrandomized trials70–74 have been largely positive, while in 5 of 7 individual randomized clinical trials (RCTs),75–81 the intervention group did not separate from placebo for treatment of depression or mania. A meta-analysis69 of RCTs in bipolar disorder showed a signal for treatment of bipolar disorder depression, but not mania. Interpretation of the responses seen in RCTs is confounded by factors including differing design of trials, compliance to study drug, composition and dose of supplements, and potential publication bias.
The Way Forward: Lessons From Migraine
We have described several ways in which altered brain AA metabolism may be important in the pathophysiology and pharmacologic or dietary treatment of bipolar disorder and have highlighted the mixed results of n-3 PUFA supplementation trials. If the brain PUFA metabolism system is important in bipolar disorder, what might account for the lack of consistent effects of dietary n-3 PUFA supplementation? One possibility is that n-3 PUFA supplementation without concurrent dietary reduction of n-6 PUFAs may not produce therapeutically relevant alterations in the interactive brain n-6 and n-3 PUFA pathways.82,83 In this regard, consumption of the n-6 PUFA precursor linoleic acid has increased in the past 100 years in the average US diet—the predictable effect of this is increasing tissue concentrations of linoleic acid and decreasing tissue concentrations of n-3 EPA and DHA, and thus an imbalance of n-6 over n-3 PUFAs and their metabolites.84 Simple addition of an n-3 PUFA supplement without concurrent reduction in dietary n-6 linoleic acid may not alter brain PUFA metabolism to the extent required to produce clinically meaningful benefit. To gain some insight into this issue and its relevance to bipolar disorder, we will describe a recent dietary intervention trial85 in migraine headache.
Migraine headache has a clinical comorbidity with bipolar disorder of around 30% for both genders when studied together,86–92 and in a recent study that investigated comorbidity by gender, 39% of women and 16% of men had migraine headache.93 The pain associated with migraine headache has been hypothesized to be caused by prostaglandin E2,94 and thus related to increased AA metabolism95 as well as increased neuroinflammation in general.96–98 Both bipolar disorder and migraine headache respond to several of the same medications, including valproic acid99,100 and lamotrigine,101 indicating a potential shared pathology.
A recent 12-week, randomized clinical trial by Ramsden et al85 compared clinical efficacy and biochemical effects of a high n-3 EPA + DHA plus low n-6 linoleic acid (H3-L6) diet to effects of only a low n-6 PUFA diet (L6) in 67 patients with chronic headache. Both the H3-L6 and L6 groups experienced statistically significant clinical improvement compared to the preintervention run-in phase, but the H3-L6 group experienced a significantly greater reduction in headache hours per day, headache days per month, headache-related quality-of-life, and psychological distress. Clinical improvements in the H3-L6 group were accompanied by reductions in erythrocyte linoleic acid and AA, as well as bioactive oxidized linoleic acid and AA metabolites that have been linked to pain.102 The H3-L6 intervention also increased n-3 EPA, DHA, and the n-3 index, as well as pathway precursors for biosynthesis of anti-inflammatory and proresolving EPA and DHA metabolites.96 Thus, the Ramsden et al trial85 suggests that lowering dietary n-6 linoleic acid may be a key component to efficacy of n-3 PUFA supplementation in migraine treatment. On the basis of the clinical and neuroinflammatory links between bipolar disorder and migraine, concurrent dietary n-6 lowering in bipolar disorder may also be necessary for effective treatment of bipolar disorder with n-3 PUFA supplementation.103
Summary and Future Directions
An extensive body of human postmortem and animal studies implicates excessive brain AA metabolism and inadequate DHA metabolism in bipolar disorder pathogenesis and progression. However, the specific molecular mechanisms linking dysfunctional AA and DHA metabolism to bipolar disorder are incompletely understood. Future studies should be directed toward identifying specific signaling pathways and lipid mediators linking AA and DHA to bipolar disorder pathophysiology. This line of inquiry could lead to development of novel, targeted strategies for affecting PUFA metabolism through modulation of dietary AA and DHA intake that can be tested for improvement of mood stabilization in randomized controlled trials.
Submitted: October 2, 2015; accepted January 11, 2016.
Potential conflicts of interest: Dr Saunders has been a consultant for Projects In Knowledge, CME, Dr Gelenberg has received an investigator-initiated grant through Penn State from Pfizer; has served as a consultant to Zynx, Allergan, Forest, and ZARS Pharma; and is a major stock owner of Healthcare Technology Systems. Drs Ramsden, Sherazy, Davis, and Rapoport have nothing to disclose.
Funding/support: The project described was supported by the National Center for Research Resources, grant KL2 RR033180 (Dr Saunders), and is now at the National Center for Advancing Translational Sciences, grant KL2 TR000126, National Institute on Aging, National Institutes of Health (NIH), Bethesda, Maryland. The contribution of Dr Rapoport was supported entirely by the Intramural Program of the National Institute on Aging, NIH, Bethesda, Maryland. The contribution of Dr Ramsden was supported by the National Institute on Alcohol Abuse and Alcoholism, NIH, Bethesda, Maryland.
Role of the sponsor: The sponsors of this research did not have direct influence over the collection, analysis or interpretation of data.
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Neither Dr Gelenberg, JCP’s editor in chief, nor Drs Saunders and Davis, editorial board members, were involved in the editorial review or decision to publish this article.
Acknowledgments: The authors would like to acknowledge Aubrey Reider, BA (Penn State College of Medicine, Hershey, Pennsylvania) for technical assistance with the figure. Ms Reider has no conflicts of interest to report.
REFERENCES
1. Goodwin FK, Jamison KR. Manic-Depressive Illness: Bipolar Disorders and Recurrent Depression. 2nd ed. New York, NY: Oxford University Press; 2007.
2. Ng F, Mammen OK, Wilting I, et al; International Society for Bipolar Disorders. The International Society for Bipolar Disorders (ISBD) consensus guidelines for the safety monitoring of bipolar disorder treatments. Bipolar Disord. 2009;11(6):559–595. doi:10.1111/j.1399-5618.2009.00737.x PubMed
3. Rapoport SI. Lithium and the other mood stabilizers effective in bipolar disorder target the rat brain arachidonic acid cascade. ACS Chem Neurosci. 2014;5(6):459–467. doi:10.1021/cn500058v PubMed
4. Rapoport SI, Basselin M, Kim HW, et al. Bipolar disorder and mechanisms of action of mood stabilizers. Brain Res Brain Res Rev. 2009;61(2):185–209. doi:10.1016/j.brainresrev.2009.06.003 PubMed
5. Rapoport SI, Bosetti F. Do lithium and anticonvulsants target the brain arachidonic acid cascade in bipolar disorder? Arch Gen Psychiatry. 2002;59(7):592–596. doi:10.1001/archpsyc.59.7.592 PubMed
6. Hibbeln JR, Palmer JW, Davis JM. Are disturbances in lipid-protein interactions by phospholipase-A2 a predisposing factor in affective illness? Biol Psychiatry. 1989;25(7):945–961. doi:10.1016/0006-3223(89)90274-6 PubMed
7. Allison JH, Stewart MA. Reduced brain inositol in lithium-treated rats. Nat New Biol. 1971;233(43):267–268. doi:10.1038/newbio233267a0 PubMed
8. Svennerholm L. Distribution and fatty acid composition of phosphoglycerides in normal human brain. J Lipid Res. 1968;9(5):570–579. PubMed
9. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi:10.1126/science.294.5548.1871 PubMed
10. Hickie I, Lloyd A. Are cytokines associated with neuropsychiatric syndromes in humans? Int J Immunopharmacol. 1995;17(8):677–683. doi:10.1016/0192-0561(95)00054-6 PubMed
11. Dantzer R, Bluthé RM, Gheusi G, et al. Molecular basis of sickness behavior. Ann N Y Acad Sci. 1998;856(1 MOLECULAR MEC):132–138. doi:10.1111/j.1749-6632.1998.tb08321.x PubMed
12. Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21(2):153–160. doi:10.1016/j.bbi.2006.09.006 PubMed
13. Parsadaniantz SM, Lebeau A, Duval P, et al. Effects of the inhibition of cyclo-oxygenase 1 or 2 or 5-lipoxygenase on the activation of the hypothalamic-pituitary-adrenal axis induced by interleukin-1beta in the male Rat. J Neuroendocrinol. 2000;12(8):766–773. doi:10.1046/j.1365-2826.2000.00517.x PubMed
14. Navarra P, Tsagarakis S, Faria MS, et al. Interleukins-1 and -6 stimulate the release of corticotropin-releasing hormone-41 from rat hypothalamus in vitro via the eicosanoid cyclooxygenase pathway. Endocrinology. 1991;128(1):37–44. doi:10.1210/endo-128-1-37 PubMed
15. Serhan CN, Arita M, Hong S, et al. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids. 2004;39(11):1125–1132. doi:10.1007/s11745-004-1339-7 PubMed
16. Nye EJ, Hockings GI, Grice JE, et al. Aspirin inhibits vasopressin-induced hypothalamic-pituitary-adrenal activity in normal humans. J Clin Endocrinol Metab. 1997;82(3):812–817. PubMed
17. Blanchard HC, Taha AY, Rapoport SI, et al. Low-dose aspirin (acetylsalicylate) prevents increases in brain PGE2, 15-epi-lipoxin A4 and 8-isoprostane concentrations in 9 month-old HIV-1 transgenic rats, a model for HIV-1 associated neurocognitive disorders. Prostaglandins Leukot Essent Fatty Acids. 2015;96:25–30. doi:10.1016/j.plefa.2015.01.002 PubMed
18. Dawson E, Parfitt E, Roberts Q, et al. Linkage studies of bipolar disorder in the region of the Darier’s disease gene on chromosome 12q23-24.1. Am J Med Genet. 1995;60(2):94–102. doi:10.1002/ajmg.1320600203 PubMed
19. Dawson E, Gill M, Curtis D, et al. Genetic association between alleles of pancreatic phospholipase A2 gene and bipolar affective disorder. Psychiatr Genet. 1995;5(4):177–180. doi:10.1097/00041444-199524000-00005 PubMed
20. Jacobsen N, Daniels J, Moorhead S, et al. Association study of bipolar disorder at the phospholipase A2 gene (PLA2A) in the Darier’s disease (DAR) region of chromosome 12q23-q24.1. Psychiatr Genet. 1996;6(4):195–199. doi:10.1097/00041444-199624000-00005 PubMed
21. Jacobsen NJ, Franks EK, Owen MJ, et al. Mutational analysis of phospholipase A2A: a positional candidate susceptibility gene for bipolar disorder. Mol Psychiatry. 1999;4(3):274–279. doi:10.1038/sj.mp.4000476 PubMed
22. Meira-Lima I, Jardim D, Junqueira R, et al. Allelic association study between phospholipase A2 genes and bipolar affective disorder. Bipolar Disord. 2003;5(4):295–299. doi:10.1034/j.1399-5618.2003.00025.x PubMed
23. Dikeos DG, Papadimitriou GN, Souery D, et al. Lack of genetic association between the phospholipase A2 gene and bipolar mood disorder in a European multicentre case-control study. Psychiatr Genet. 2006;16(4):169–171. doi:10.1097/01.ypg.0000218615.19892.86 PubMed
24. Noponen M, Sanfilipo M, Samanich K, et al. Elevated PLA2 activity in schizophrenics and other psychiatric patients. Biol Psychiatry. 1993;34(9):641–649. doi:10.1016/0006-3223(93)90157-9 PubMed
25. Ross BM, Hughes B, Kish SJ, et al. Serum calcium-independent phospholipase A2 activity in bipolar affective disorder. Bipolar Disord. 2006;8(3):265–270. doi:10.1111/j.1399-5618.2006.00299.x PubMed
26. Ikenaga EH, Talib LL, Ferreira AS, et al. Reduced activities of phospholipases A2 in platelets of drug-naive bipolar disorder patients. Bipolar Disord. 2015;17(1):97–101. doi:10.1111/bdi.12229 PubMed
27. Kim HW, Rapoport SI, Rao JS. Altered arachidonic acid cascade enzymes in postmortem brain from bipolar disorder patients. Mol Psychiatry. 2011;16(4):419–428. doi:10.1038/mp.2009.137 PubMed
28. Lee HJ, Rao JS, Chang L, et al. Chronic N-methyl-d-aspartate administration increases the turnover of arachidonic acid within brain phospholipids of the unanesthetized rat. J Lipid Res. 2008;49(1):162–168. doi:10.1194/jlr.M700406-JLR200 PubMed
29. Rao JS, Ertley RN, Rapoport SI, et al. Chronic NMDA administration to rats up-regulates frontal cortex cytosolic phospholipase A2 and its transcription factor, activator protein-2. J Neurochem. 2007;102(6):1918–1927. doi:10.1111/j.1471-4159.2007.04648.x PubMed
30. Kim HW, Rapoport SI, Rao JS. Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol Dis. 2010;37(3):596–603. doi:10.1016/j.nbd.2009.11.010 PubMed
31. Brock TG. Arachidonic acid binds 14-3-3zeta, releases 14-3-3zeta from phosphorylated BAD and induces aggregation of 14-3-3zeta. Neurochem Res. 2008;33(5):801–807. doi:10.1007/s11064-007-9498-3 PubMed
32. Rao JS, Harry GJ, Rapoport SI, et al. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry. 2010;15(4):384–392. doi:10.1038/mp.2009.47 PubMed
33. Saunders EFH, Ramsden CE, Sherazy MS, et al. Omega-3 and omega-6 polyunsaturated fatty acids in bipolar disorder: a review of biomarker and treatment studies. J Clin Psychiatry. 2016;77(10):e1301–e1308.
34. Saunders EF, Reider A, Singh G, et al. Low unesterified:esterified eicosapentaenoic acid (EPA) plasma concentration ratio is associated with bipolar disorder episodes, and omega-3 plasma concentrations are altered by treatment. Bipolar Disord. 2015;17(7):729–742. doi:10.1111/bdi.12337 PubMed
35. Chiu CC, Huang SY, Su KP, et al. Polyunsaturated fatty acid deficit in patients with bipolar mania. Eur Neuropsychopharmacol. 2003;13(2):99–103. doi:10.1016/S0924-977X(02)00130-X PubMed
36. Sublette ME, Bosetti F, DeMar JC, et al. Plasma free polyunsaturated fatty acid levels are associated with symptom severity in acute mania. Bipolar Disord. 2007;9(7):759–765. doi:10.1111/j.1399-5618.2007.00387.x PubMed
37. Clayton EH, Hanstock TL, Hirneth SJ, et al. Long-chain omega-3 polyunsaturated fatty acids in the blood of children and adolescents with juvenile bipolar disorder. Lipids. 2008;43(11):1031–1038. doi:10.1007/s11745-008-3224-z PubMed
38. McNamara RK, Jandacek R, Rider T, et al. Selective deficits in erythrocyte docosahexaenoic acid composition in adult patients with bipolar disorder and major depressive disorder. J Affect Disord. 2010;126(1–2):303–311. doi:10.1016/j.jad.2010.03.015 PubMed
39. Evans SJ, Kamali M, Prossin AR, et al. Association of plasma ω-3 and ω-6 lipids with burden of disease measures in bipolar subjects. J Psychiatr Res. 2012;46(11):1435–1441. doi:10.1016/j.jpsychires.2012.07.016 PubMed
40. Pomponi M, Janiri L, La Torre G, et al. Plasma levels of n-3 fatty acids in bipolar patients: deficit restricted to DHA. J Psychiatr Res. 2013;47(3):337–342. doi:10.1016/j.jpsychires.2012.11.004 PubMed
41. Berk M, Kapczinski F, Andreazza AC, et al. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev. 2011;35(3):804–817. doi:10.1016/j.neubiorev.2010.10.001 PubMed
42. Vederman AC, Weisenbach SL, Rapport LJ, et al. Modality-specific alterations in the perception of emotional stimuli in bipolar disorder compared to healthy controls and major depressive disorder. Cortex. 2012;48(8):1027–1034. doi:10.1016/j.cortex.2011.03.017 PubMed
43. Ryan KA, Vederman AC, McFadden EM, et al. Differential executive functioning performance by phase of bipolar disorder. Bipolar Disord. 2012;14(5):527–536. doi:10.1111/j.1399-5618.2012.01032.x PubMed
44. Langenecker SA, Saunders EF, Kade AM, et al. Intermediate: cognitive phenotypes in bipolar disorder. J Affect Disord. 2010;122(3):285–293. doi:10.1016/j.jad.2009.08.018 PubMed
45. Haarman BC, Riemersma-Van der Lek RF, Burger H, et al. Relationship between clinical features and inflammation-related monocyte gene expression in bipolar disorder—towards a better understanding of psychoimmunological interactions. Bipolar Disord. 2014;16(2):137–150. doi:10.1111/bdi.12142 PubMed
46. Becking K, Boschloo L, Vogelzangs N, et al. The association between immune activation and manic symptoms in patients with a depressive disorder. Transl Psychiatr. 2013;3(10):e314. doi:10.1038/tp.2013.87 PubMed
47. Bora E, Yücel M, Pantelis C, et al. Meta-analytic review of neurocognition in bipolar II disorder. Acta Psychiatr Scand. 2011;123(3):165–174. doi:10.1111/j.1600-0447.2010.01638.x PubMed
48. DelBello MP, Strakowski SM, Zimmerman ME, et al. MRI analysis of the cerebellum in bipolar disorder: a pilot study. Neuropsychopharmacology. 1999;21(1):63–68. doi:10.1016/S0893-133X(99)00026-3 PubMed
49. Lyoo IK, Sung YH, Dager SR, et al. Regional cerebral cortical thinning in bipolar disorder. Bipolar Disord. 2006;8(1):65–74. doi:10.1111/j.1399-5618.2006.00284.x PubMed
50. Savitz J, Frank MB, Victor T, et al. Inflammation and neurological disease-related genes are differentially expressed in depressed patients with mood disorders and correlate with morphometric and functional imaging abnormalities. Brain Behav Immun. 2013;31:161–171. doi:10.1016/j.bbi.2012.10.007 PubMed
51. Savitz J, Dantzer R, Wurfel BE, et al. Neuroprotective kynurenine metabolite indices are abnormally reduced and positively associated with hippocampal and amygdalar volume in bipolar disorder. Psychoneuroendocrinology. 2015;52:200–211. doi:10.1016/j.psyneuen.2014.11.015 PubMed
52. Haarman BC, Burger H, Doorduin J, et al. Volume, metabolites and neuroinflammation of the hippocampus in bipolar disorder—a combined magnetic resonance imaging and positron emission tomography study. Brain Behav Immun. 2016;56:21–33. doi:10.1016/j.bbi.2015.09.004 PubMed
53. Garrido R, Springer JE, Hennig B, et al. Apoptosis of spinal cord neurons by preventing depletion nicotine attenuates arachidonic acid-induced of neurotrophic factors. J Neurotrauma. 2003;20(11):1201–1213. doi:10.1089/089771503322584628 PubMed
54. Chang MC, Bell JM, Purdon AD, et al. Dynamics of docosahexaenoic acid metabolism in the central nervous system: lack of effect of chronic lithium treatment. Neurochem Res. 1999;24(3):399–406. doi:10.1023/A:1020989701330 PubMed
55. Bazinet RP, Weis MT, Rapoport SI, et al. Valproic acid selectively inhibits conversion of arachidonic acid to arachidonoyl-CoA by brain microsomal long-chain fatty acyl-CoA synthetases: relevance to bipolar disorder. Psychopharmacology (Berl). 2006;184(1):122–129. doi:10.1007/s00213-005-0272-4 PubMed
56. Bazinet RP, Rao JS, Chang L, et al. Chronic carbamazepine decreases the incorporation rate and turnover of arachidonic acid but not docosahexaenoic acid in brain phospholipids of the unanesthetized rat: relevance to bipolar disorder. Biol Psychiatry. 2006;59(5):401–407. doi:10.1016/j.biopsych.2005.07.024 PubMed
57. Lee HJ, Ertley RN, Rapoport SI, et al. Chronic administration of lamotrigine downregulates COX-2 mRNA and protein in rat frontal cortex. Neurochem Res. 2008;33(5):861–866. doi:10.1007/s11064-007-9526-3 PubMed
58. Lee HJ, Ghelardoni S, Chang L, et al. Topiramate does not alter the kinetics of arachidonic or docosahexaenoic acid in brain phospholipids of the unanesthetized rat. Neurochem Res. 2005;30(5):677–683. doi:10.1007/s11064-005-2756-3 PubMed
59. Ghelardoni S, Bazinet RP, Rapoport SI, et al. Topiramate does not alter expression in rat brain of enzymes of arachidonic acid metabolism. Psychopharmacology (Berl). 2005;180(3):523–529. doi:10.1007/s00213-005-2189-3 PubMed
60. Reese EA, Cheon Y, Ramadan E, et al. Gabapentin’s minimal action on markers of rat brain arachidonic acid metabolism agrees with its inefficacy against bipolar disorder. Prostaglandins Leukot Essent Fatty Acids. 2012;87(2–3):71–77. doi:10.1016/j.plefa.2012.06.003 PubMed
61. Stolk P, Souverein PC, Wilting I, et al. Is aspirin useful in patients on lithium? a pharmacoepidemiological study related to bipolar disorder. Prostaglandins Leukot Essent Fatty Acids. 2010;82(1):9–14. doi:10.1016/j.plefa.2009.10.007 PubMed
62. Nery FG, Monkul ES, Hatch JP, et al. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol. 2008;23(2):87–94. doi:10.1002/hup.912 PubMed
63. Pacchiarotti I, Bond DJ, Baldessarini RJ, et al. The International Society for Bipolar Disorders (ISBD) task force report on antidepressant use in bipolar disorders. Am J Psychiatry. 2013;170(11):1249–1262. doi:10.1176/appi.ajp.2013.13020185 PubMed
64. Lee HJ, Rao JS, Chang L, et al. Chronic imipramine but not bupropion increases arachidonic acid signaling in rat brain: is this related to ‘switching’ in bipolar disorder? Mol Psychiatry. 2010;15(6):602–614. doi:10.1038/mp.2008.117 PubMed
65. Rao JS, Ertley RN, Lee HJ, et al. Chronic fluoxetine upregulates activity, protein and mRNA levels of cytosolic phospholipase A2 in rat frontal cortex. Pharmacogenomics J. 2006;6(6):413–420. doi:10.1038/sj.tpj.6500391 PubMed
66. Edwards SJ, Hamilton V, Nherera L, et al. Lithium or an atypical antipsychotic drug in the management of treatment-resistant depression: a systematic review and economic evaluation. Health Technol Assess. 2013;17(54):1–190. doi:10.3310/hta17540 PubMed
67. Warner-Schmidt JL, Vanover KE, Chen EY, et al. Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs) are attenuated by antiinflammatory drugs in mice and humans. Proc Natl Acad Sci U S A. 2011;108(22):9262–9267. doi:10.1073/pnas.1104836108 PubMed
68. Su KP, Lai HC, Yang HT, et al. Omega-3 fatty acids in the prevention of interferon-alpha-induced depression: results from a randomized, controlled trial. Biol Psychiatry. 2014;76(7):559–566. doi:10.1016/j.biopsych.2014.01.008 PubMed
69. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81–86. doi:10.4088/JCP.10r06710 PubMed
70. Hirashima F, Parow AM, Stoll AL, et al. Omega-3 fatty acid treatment and T(2) whole brain relaxation times in bipolar disorder. Am J Psychiatry. 2004;161(10):1922–1924. doi:10.1176/ajp.161.10.1922 PubMed
71. Osher Y, Bersudsky Y, Belmaker RH. Omega-3 eicosapentaenoic acid in bipolar depression: report of a small open-label study. J Clin Psychiatry. 2005;66(6):726–729. doi:10.4088/JCP.v66n0608 PubMed
72. Sagduyu K, Dokucu ME, Eddy BA, et al. Omega-3 fatty acids decreased irritability of patients with bipolar disorder in an add-on, open label study. Nutr J. 2005;4(1):6. doi:10.1186/1475-2891-4-6 PubMed
73. Wozniak J, Biederman J, Mick E, et al. Omega-3 fatty acid monotherapy for pediatric bipolar disorder: a prospective open-label trial. Eur Neuropsychopharmacol. 2007;17(6–7):440–447. doi:10.1016/j.euroneuro.2006.11.006 PubMed
74. Clayton EH, Hanstock TL, Hirneth SJ, et al. Reduced mania and depression in juvenile bipolar disorder associated with long-chain omega-3 polyunsaturated fatty acid supplementation. Eur J Clin Nutr. 2009;63(8):1037–1040. doi:10.1038/ejcn.2008.81 PubMed
75. Stoll AL, Severus WE, Freeman MP, et al. Omega 3 fatty acids in bipolar disorder: a preliminary double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1999;56(5):407–412. doi:10.1001/archpsyc.56.5.407 PubMed
76. Chiu CC, Huang SY, Chen CC, et al. Omega-3 fatty acids are more beneficial in the depressive phase than in the manic phase in patients with bipolar I disorder. J Clin Psychiatry. 2005;66(12):1613–1614. doi:10.4088/JCP.v66n1219b PubMed
77. Keck PE Jr, Mintz J, McElroy SL, et al. Double-blind, randomized, placebo-controlled trials of ethyl-eicosapentanoate in the treatment of bipolar depression and rapid cycling bipolar disorder. Biol Psychiatry. 2006;60(9):1020–1022. doi:10.1016/j.biopsych.2006.03.056 PubMed
78. Frangou S, Lewis M, McCrone P. Efficacy of ethyl-eicosapentaenoic acid in bipolar depression: randomised double-blind placebo-controlled study. Br J Psychiatry. 2006;188(1):46–50. doi:10.1192/bjp.188.1.46 PubMed
79. Frangou S, Lewis M, Wollard J, et al. Preliminary in vivo evidence of increased N-acetyl-aspartate following eicosapentanoic acid treatment in patients with bipolar disorder. J Psychopharmacol. 2007;21(4):435–439. doi:10.1177/0269881106067787 PubMed
80. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142–154. doi:10.1111/j.1399-5618.2010.00799.x PubMed
81. Murphy BL, Stoll AL, Harris PQ, et al. Omega-3 fatty acid treatment, with or without cytidine, fails to show therapeutic properties in bipolar disorder: a double-blind, randomized add-on clinical trial. J Clin Psychopharmacol. 2012;32(5):699–703. doi:10.1097/JCP.0b013e318266854c PubMed
82. Rapoport SI. Brain arachidonic and docosahexaenoic acid cascades are selectively altered by drugs, diet and disease. Prostaglandins Leukot Essent Fatty Acids. 2008;79(3–5):153–156. doi:10.1016/j.plefa.2008.09.010 PubMed
83. Ryan VH, Primiani CT, Rao JS, et al. Coordination of gene expression of arachidonic and docosahexaenoic acid cascade enzymes during human brain development and aging. PLoS ONE. 2014;9(6):e100858. doi:10.1371/journal.pone.0100858 PubMed
84. Blasbalg TL, Hibbeln JR, Ramsden CE, et al. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr. 2011;93(5):950–962. doi:10.3945/ajcn.110.006643 PubMed
85. Ramsden CE, Faurot KR, Zamora D, et al. Targeted alteration of dietary n-3 and n-6 fatty acids for the treatment of chronic headaches: a randomized trial. Pain. 2013;154(11):2441–2451. doi:10.1016/j.pain.2013.07.028 PubMed
86. Baptista T, Uzcátegui E, Arapé Y, et al. Migraine life-time prevalence in mental disorders: concurrent comparisons with first-degree relatives and the general population.
Invest Clin. 2012;53(1):38–51. PubMed
87. Ortiz A, Cervantes P, Zlotnik G, et al. Cross-prevalence of migraine and bipolar disorder. Bipolar Disord. 2010;12(4):397–403. doi:10.1111/j.1399-5618.2010.00832.x PubMed
88. Dilsaver SC, Benazzi F, Oedegaard KJ, et al. Migraine headache in affectively ill Latino adults of Mexican American origin is associated with bipolarity. Prim Care Companion J Clin Psychiatry. 2009;11(6):302–306. doi:10.4088/PCC.08m00728 PubMed
89. McIntyre RS, Konarski JZ, Soczynska JK, et al. Medical comorbidity in bipolar disorder: implications for functional outcomes and health service utilization. Psychiatr Serv. 2006;57(8):1140–1144. doi:10.1176/ps.2006.57.8.1140 PubMed
90. McIntyre RS, Konarski JZ, Wilkins K, et al. The prevalence and impact of migraine headache in bipolar disorder: results from the Canadian Community Health Survey. Headache. 2006;46(6):973–982. doi:10.1111/j.1526-4610.2006.00469.x PubMed
91. Fasmer OB. The prevalence of migraine in patients with bipolar and unipolar affective disorders. Cephalalgia. 2001;21(9):894–899. doi:10.1046/j.1468-2982.2001.00279.x PubMed
92. Fasmer OB, Oedegaard KJ. Clinical characteristics of patients with major affective disorders and comorbid migraine. World J Biol Psychiatry. 2001;2(3):149–155. doi:10.3109/15622970109026801 PubMed
93. Saunders EF, Nazir R, Kamali M, et al. Gender differences, clinical correlates, and longitudinal outcome of bipolar disorder with comorbid migraine. J Clin Psychiatry. 2014;75(5):512–519. doi:10.4088/JCP.13m08623 PubMed
94. Antonova M, Wienecke T, Olesen J, et al. Prostaglandin E(2) induces immediate migraine-like attack in migraine patients without aura. Cephalalgia. 2012;32(11):822–833. doi:10.1177/0333102412451360 PubMed
95. Ramsden CE, Mann JD, Faurot KR, et al. Low omega-6 vs low omega-6 plus high omega-3 dietary intervention for chronic daily headache: protocol for a randomized clinical trial. Trials. 2011;12(1):97. doi:10.1186/1745-6215-12-97 PubMed
96. Moskowitz MA, Buzzi MG. Migraine general aspects. Handb Clin Neurol. 2010;97:253–266. doi:10.1016/S0072-9752(10)97021-8 PubMed
97. Buzzi MG, Moskowitz MA. The pathophysiology of migraine: year 2005. J Headache Pain. 2005;6(3):105–111. doi:10.1007/s10194-005-0165-2 PubMed
98. Buzzi MG, Moskowitz MA. The trigemino-vascular system and migraine. Pathol Biol (Paris). 1992;40(4):313–317. PubMed
99. Geddes JR, Goodwin GM, Rendell J, et al; BALANCE investigators and collaborators. Lithium plus valproate combination therapy versus monotherapy for relapse prevention in bipolar I disorder (BALANCE): a randomised open-label trial. Lancet. 2010;375(9712):385–395. doi:10.1016/S0140-6736(09)61828-6 PubMed
100. Linde M, Mulleners WM, Chronicle EP, et al. Valproate (valproic acid or sodium valproate or a combination of the two) for the prophylaxis of episodic migraine in adults. Cochrane Database Syst Rev. 2013;6(6):CD010611. PubMed
101. Lampl C, Katsarava Z, Diener HC, et al. Lamotrigine reduces migraine aura and migraine attacks in patients with migraine with aura. J Neurol Neurosurg Psychiatry. 2005;76(12):1730–1732. doi:10.1136/jnnp.2005.063750 PubMed
102. Ramsden CE, Ringel A, Feldstein AE, et al. Lowering dietary linoleic acid reduces bioactive oxidized linoleic acid metabolites in humans. Prostaglandins Leukot Essent Fatty Acids. 2012;87(4–5):135–141. doi:10.1016/j.plefa.2012.08.004 PubMed
103. MacIntosh BA, Ramsden CE, Faurot KR, et al. Low-n-6 and low-n-6 plus high-n-3 diets for use in clinical research. Br J Nutr. 2013;110(3):559–568. doi:10.1017/S0007114512005181 PubMed
This PDF is free for all visitors!



