Click to enlarge page
Clinicians are often faced with depressed patients who do not respond to treatment or who continue to experience troublesome residual symptoms. For these patients, medications with different mechanisms of action may improve symptoms. By altering levels of dopamine, norepinephrine, and serotonin in the brain, clinicians theoretically may be able to target specific depressive symptoms, but they must be aware of the associated adverse effects. Follow a discussion by expert faculty as they review the mechanisms of action of the different antidepressant classes from the older tricyclic antidepressants and monoamine oxidase inhibitors to the newer selective serotonin reuptake inhibitors and dual-acting agents.
From the Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia (Dr Thase) and the Department of Psychiatry, SUNY Upstate Medical University, Syracuse, New York (Dr Schwartz).
This CME activity is expired. For more CME activities, visit CMEInstitute.com.
Find more articles on this and other psychiatry and CNS topics:
The Journal of Clinical Psychiatry
The Primary Care Companion for CNS Disorders

This Academic Highlights section of The Journal of Clinical Psychiatry presents the highlights of the planning teleconference series “Using Mechanism of Action to Choose Medications for Treatment-Resistant Depression,” which was held in September and October 2014. This report was prepared and independently developed by the CME Institute of Physicians Postgraduate Press, Inc., and was supported by an educational grant from Otsuka America Pharmaceutical, Inc.
The teleconference was chaired by Michael E. Thase, MD, Department of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia. The faculty was Thomas L. Schwartz, MD, Department of Psychiatry, SUNY Upstate Medical University, Syracuse, New York.
Financial disclosure: Dr Thase has been an advisor/consultant for Alkermes, Allergan, AstraZeneca, Bristol-Myers Squibb, Cerecor, Eli Lilly, Forest, Gerson Lehrman, GlaxoSmithKline, Guidepoint Global, Lundbeck, MedAvante, Merck, Moksha8, Neuronetics, Novartis, Ortho-McNeil, Otsuka, Pamlab, Pfizer, PGx, Shire, Sunovion, and Takeda; has received grant/research support from the Agency for Healthcare Research and Quality, Forest, National Institute of Mental Health, and Otsuka; has equity holdings in MedAvante; and has received royalties from the American Psychiatric Foundation, Guilford Publications, Herald House, and W. W. Norton & Company; his spouse/partner is employed by Peloton Advantage. Dr Schwartz has received grant/research support from Cyberonics.
The opinions expressed herein are those of the faculty and do not necessarily reflect the opinions of the CME provider and publisher or the commercial supporter.
J Clin Psychiatry 2015;76(6):720-727 (doi:10.4088/JCP.14052ah2c)
© Copyright 2015 Physicians Postgraduate Press, Inc.
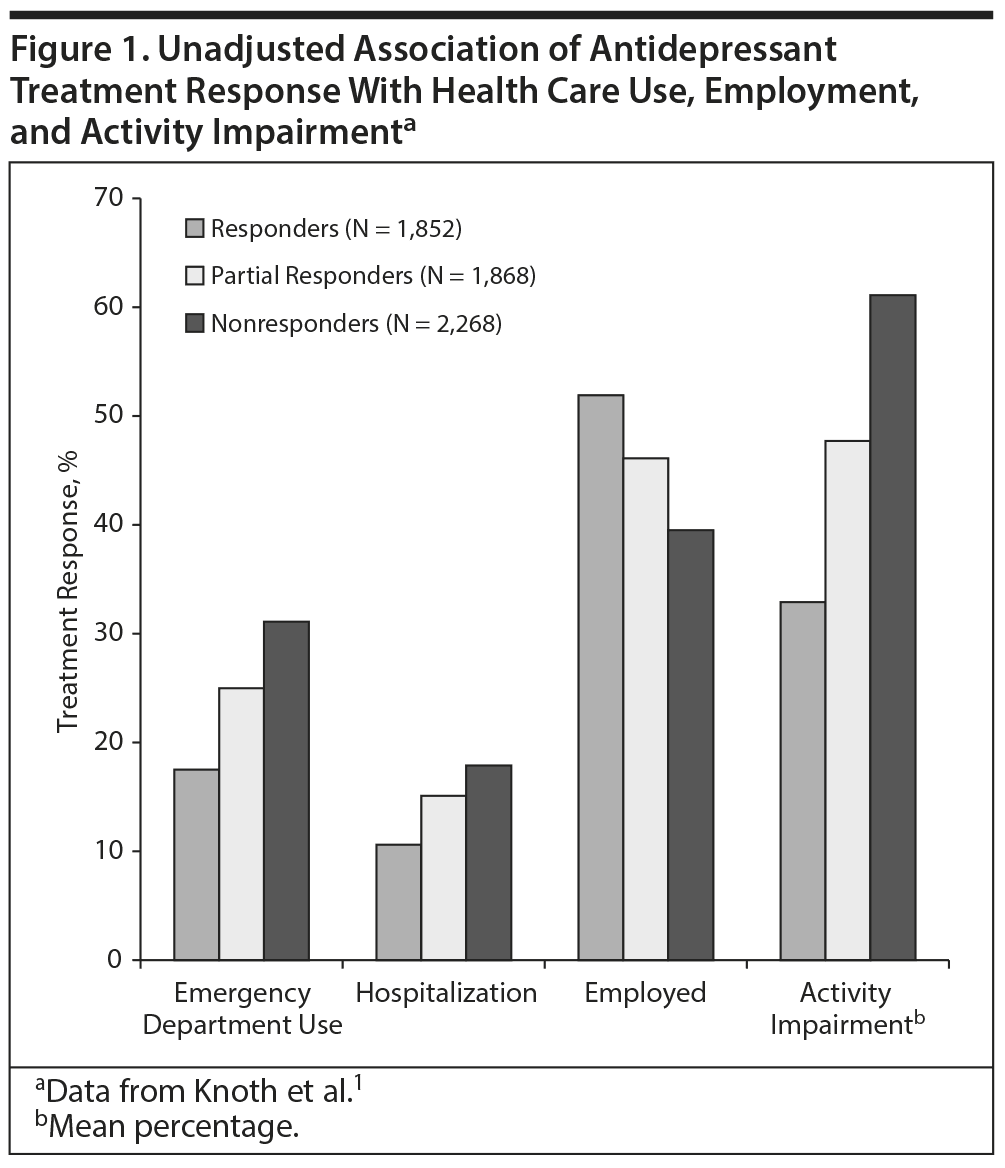
Despite the growing number of antidepressants approved by the US Food and Drug Administration (FDA), many patients with depression fail to respond to treatment, while others improve but experience residual symptoms that affect their daily function. In a study1 that analyzed data from a self-report survey, 5,988 adult participants met inclusion criteria of a major depressive disorder (MDD) diagnosis and current antidepressant treatment. Of these participants, 30.9% responded to treatment and 31.2% experienced partial response while 37.9% did not respond. Participants who failed to respond used more health care resources, were less likely to be employed, and were less productive at work than those who responded to treatment (Figure 1).1 In a cohort of 930 outpatients with MDD, 624 patients (67%) had responded after 3 months of naturalistic treatment,2 meaning that one-third had not responded. Among the responders, 412 patients (66%) had reached remission. However, among those in remission, 88% had residual symptoms including anxiety, core mood symptoms, insomnia, somatic problems, and pain, which were associated with different degrees of reduced functioning.2
Click figure to enlarge
Patients are typically considered to have treatment-resistant depression if they fail to achieve remission after at least 2 trials of antidepressants from different pharmacologic classes.1 Although antidepressants generally show similar efficacy in clinical trials,3 they have different mechanisms of action (MOA) that influence how well they may work for individual patients based on particular symptom constellations. To achieve better outcomes for their patients with depression, clinicians should consider the MOA associated with different medication classes. Join faculty members Michael E. Thase, MD, and Thomas L. Schwartz, MD, as they outline the importance of the MOA of antidepressants, the treatment implications of the monoamine hypothesis, and the specific MOA and related adverse effects of antidepressant classes.
Importance of Mechanism of Action
Dr Thase noted that depression is a heterogeneous condition, with subgroups of patients differing in symptom syndromes and patterns over time.4 Depression is partly an elaboration of normal human responses to stress and life events,4 but etiologic factors may vary among subtypes of depression. Depression may differ among individuals in part due to malfunctions in different brain areas,5 and antidepressants affect the brain in different ways. When clinicians understand the pathways affected by different antidepressants, they can increase their ability to help patients with difficult-to-treat depression.
- The mechanism of action of antidepressants will influence their therapeutic benefits and adverse effects.
- Clinicians theoretically can match patients’ symptoms to dysfunction in dopamine, norepinephrine, and serotonin pathways and then select a medication to improve that function.
- Each antidepressant class, from TCAs and MAOIs to SSRIs, SNRIs, and beyond, has a unique mechanism of action that may be beneficial for subgroups of patients, such as those with atypical, melancholic, or treatment-resistant depression.
All antidepressants build upon the placebo response, which in clinical trials is about 30%-40%.6 Beyond their efficacy as active placebos, antidepressants have specific actions that help determine which agent may be more beneficial for a particular subgroup of depressed patients. The side effects of antidepressants, which are related to MOA just as efficacy is, can be beneficial, depending on a patient’s symptoms. For example, an antidepressant that causes sedation can help patients who are having trouble sleeping, but that side effect may be unwelcome in a patient who is already oversleeping.
Dr Schwartz stated that clinicians often learn about serotonin, norepinephrine, dopamine, and their respective receptors as the molecular entities that have been manipulated since the 1950s in order to treat depressive disorders. By selecting a medication based on what it theoretically does in the brain (both in terms of therapeutic benefit and adverse effects), clinicians theoretically can match medications to their patients’ symptoms.
Dr Thase agreed that consideration of the monoamine pathways is a good starting point. Many of the original classes of antidepressants interacted with etiologic pathways of depression by facilitating or improving serotonin or norepinephrine neurotransmission.7 Medications that primarily modulate serotonin are thought to be beneficial for dampening excessive negative emotional responses. Medications that have a predominantly noradrenergic MOA may be better at improving positive affect, energy, and concentration. Primarily dopaminergic antidepressants are currently not available, but dopamine has important effects on drive, reward, or consummatory motivation.5 Most antidepressants are primarily serotonergic or noradrenergic with only indirect dopaminergic neurotransmission, which may explain why symptoms such as impaired motivation and pleasure remain in some patients with depression.8 Triple reuptake inhibitors, which inhibit the reuptake of all 3 monoamines including dopamine, have a chance to provide greater efficacy than traditional antidepressants, especially in patients with atypical and melancholic depression.8 Triple reuptake inhibitors are currently being investigated and may provide a useful addition to MDD treatments.9,10
Monoamine Hypothesis
The classic monoamine hypothesis, which has been refined over the past few decades,11 stated that when monoamine activity is normal, no depression is present, but if serotonin or norepinephrine are reduced, depressive symptoms emerge.5 Thus, antidepressants could improve symptoms of depression by increasing synaptic concentrations of these monoamines.11
Dr Schwartz explained that the current monoamine hypothesis posits that alterations in dopamine, norepinephrine, and serotonin receptors in the brain lead to depression.5,11 Specifically, high concentrations of receptors may cause depression. Dr Thase added that the classic monoamine hypothesis was created before the roles of dopamine, glutamate, and certain neuropeptides in depression were recognized.11 Because the original antidepressants enhanced or restored norepinephrine or serotonin levels, optimism was high in the 1960s and early 1970s that the cause of depression had been found. The pathophysiology of depression is now understood to be even more complex, and the monoamine hypothesis cannot provide a complete explanation for the action of antidepressants.11 However, the 3 key monoamine pathways of dopamine, norepinephrine, and serotonin in the brain are useful to study because of their potential connections with symptoms of MDD (Table 1).5
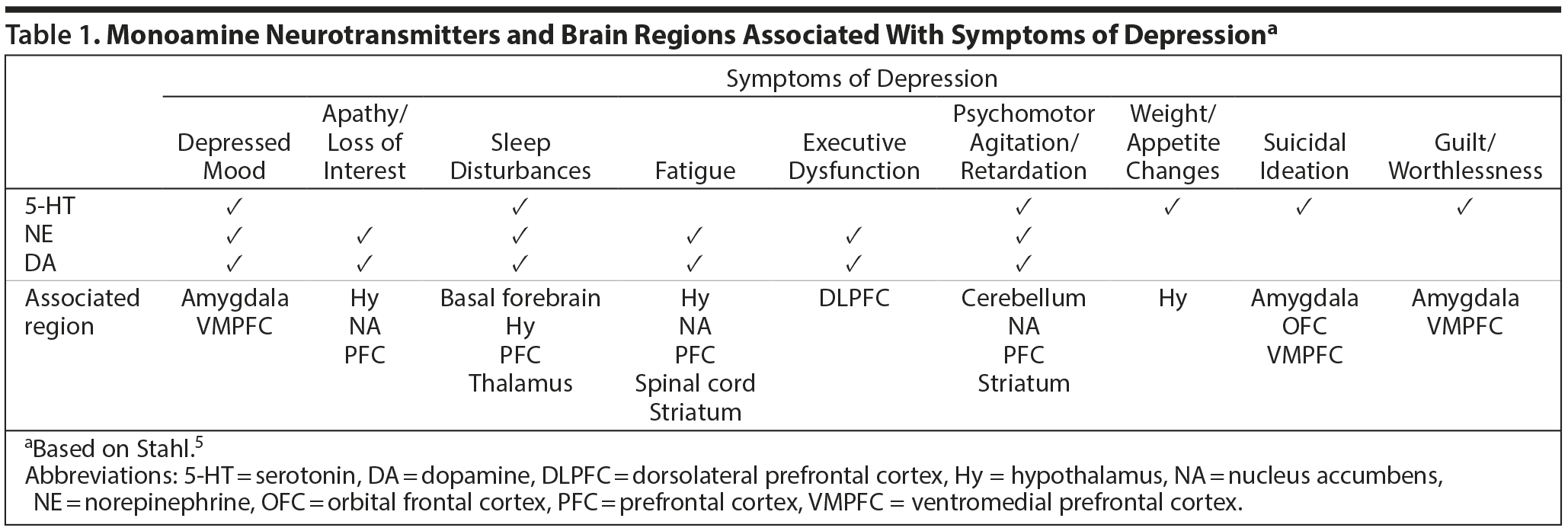
Click figure to enlarge
Dopamine
Dopamine has a couple of key pathways in the brain and is related to vigilance, alertness, drive, and motivation.5 Theoretically, dopamine dysfunction is associated with patients who are predominantly amotivated or disinterested.5 Dr Thase commented that a reduction in the activity of the nucleus accumbens, which can result in apathy, may be the product of a sustained, unresolvable stress. That is, if someone has an overwhelming problem that seems difficult to solve, and the consequences of not solving it are great, then that person has no immediate need to find fun or rewarding activities.
Some medications have more dopamine potential than others. Among the original antidepressants, the monoamine oxidase inhibitors (MAOIs) were the most dopaminergic because their MOA prevents the degradation of dopamine in the synapse.5 Among modern antidepressants, bupropion is the most dopaminergic, although positron emission tomography scans of patients taking bupropion show a relatively modest dopamine effect.5 The serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine has a modest effect on dopamine at very high doses, and the selective serotonin reuptake inhibitor (SSRI) sertraline, at high doses, can inhibit dopamine reuptake as well.12 For a more focused dopamine effect, either a psychostimulant or a second-generation antipsychotic, such as low-dose aripiprazole, is needed (Case 1).13
Case 1
Mr P, a 69-year-old widower, presented to his primary care provider with insomnia, restlessness, irritability, weight loss, concentration problems, and constipation. He denied being suicidal but asked if life was worth living since he had made a business mistake that cost him $100,000. He manages rental properties and recently refurbished a house that will not sell. His children confirm that this event precipitated his mood change, although his other business ventures are doing well. He is now oppositional, negative, and angry.
Mr P had no psychiatric history and showed no apparent psychosis, mania, anxiety disorder, eating disorder, or substance use disorder. He met Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, (DSM-5) criteria for MDD, but he showed no response to 2 adequately dosed trials of escitalopram and duloxetine. He also failed to improve after being referred to a psychiatrist, who prescribed desvenlafaxine titrated to 200 mg/d. The psychiatrist next combined desvenlafaxine with mirtazapine titrated to 45 mg/d.
At his follow-up visit, Mr P was irritable and constricted and continued to complain about his past financial failure, medication copays, and constipation problems. Suspecting that he may have fixed delusional beliefs, the psychiatrist discontinued mirtazapine and added low-dose aripiprazole.
Although the 5-mg and 10-mg daily doses of aripiprazole did not help, once it was titrated to 15 mg/d, Mr P seemed less tense and anxious and slept better. At 20 mg/d, he admitted that he “was not a financial failure” and that he had a “single business loss and not a complete business failure.” He recognized that he had plenty of money and could retire without problems. He stopped commenting about his bowel problems and medication copays. He was symptom free after a few weeks of taking the 20-mg/d dose in combination with desvenlafaxine 200 mg/d.
Mr P’s depression did not respond until aripiprazole was increased to a dose used to treat psychosis. Clinically, he had fixed beliefs that could not be dispelled by actual facts. His final diagnosis was MDD, single episode, severe, with psychotic features. He initially seemed to be in a treatment-resistant depression, but his fairly robust response to aripiprazole augmentation indicates the presence of psychotic features with MDD. If his fixed beliefs had been classified as psychotic delusions earlier, he may have experienced faster symptom resolution.
The use of 2 antidepressants, both of which increase norepinephrine and serotonin by different mechanisms, was not a strong enough combination to help Mr P. Although clinicians often do not know the exact reason that a particular medication strategy does not work for a specific patient, one possibility to consider in this case is that the patient had a form of affective illness that does not commonly respond to antidepressant medications. In this case, the working hypothesis is that Mr P had a psychotic illness that warranted an antipsychotic medication. Instead of boosting dopamine activity, it was necessary to block D2 receptors to halt the psychotic elements of his depression to allow a full antidepressant response to occur.
None of the second-generation antipsychotics are specifically approved for the treatment of psychotic forms of MDD. Nevertheless, it has been known since the 1980s that psychotic forms of MDD are more likely to respond to treatment regimens that include an antipsychotic medication than to antidepressant monotherapy. There is scant, but some, evidence to suggest that psychotic depression can respond to SSRIs in a minority of patients.14 This patient did not respond to monotherapy that included serotonin reuptake inhibition, suggesting that serotonin elevations were not the main cause of his depression and certainly not the cause of his psychosis. Psychosis is theoretically associated with elevations in dopamine activity in the limbic system.
Norepinephrine
Norepinephrine is associated with symptoms including low energy and concentration problems (see Table 1). Norepinephrine production starts in the midbrain and travels to different parts of the brain. Reduced norepinephrine, especially in the prefrontal cortex, can cause problems with alertness, concentration, decision-making, and information processing.5 These symptoms appear in patients with attention-deficit/hyperactivity disorder and also in patients with depression, who may have trouble focusing, making decisions, and completing tasks.
To increase norepinephrine, several medications are available. The tricyclic antidepressants (TCAs) are primarily noradrenergic drugs.5 For most TCAs, the ability to block the reuptake of norepinephrine is more selective than their effect on serotonin.5 Newer antidepressant drugs that have noradrenergic effects include bupropion and the SNRIs.
Serotonin
The third monoamine pathway is serotonin. Patients with serotonin dysfunction may be sad, weepy, guilt-ridden, or suicidal (see Table 1).5 Serotonin pathways are co-localized with norepinephrine. A patient with strong negative affect may have insufficient serotonergic inhibition to dampen negativity. Many available agents can enhance serotonergic activity, the most widely used being the SSRIs.
MDD Symptomatology and Brain Regions
Dr Schwartz examined how the 9 diagnostic symptoms of MDD theoretically coincide with the 3 monoamine pathways. Depression is a heterogeneous disorder, and patients may have very different presentations. Other than the core symptoms of depressed mood and/or decreased pleasure or interest in activities, the remaining symptoms can appear in various constellations. One patient may be sad, weepy, and suicidal while another has low energy, fatigue, and the inability to focus or concentrate.
Depression affects many areas of the brain. The frontal lobe is associated with energy, concentration, executive function, and the ability to focus. The negative affect or emotional aspect of depression—suicidality, guilt, and sadness—seems to reside deeper in the limbic structures of the brain.5
Dr Thase mentioned that neuroimaging studies show areas of the brain that are overfunctioning or underfunctioning in people with depression.15,16 Not all depressed people show hypofunction in the dorsolateral prefrontal cortex, and not all depressed people show hyperfunction in limbic structures such as the amygdala. But, these 2 areas are most often implicated in people who are depressed,15 with one or the other being more dominant and causing more symptoms.
Dr Schwartz wondered if, in the future, clinicians may be able to combine a thorough clinical evaluation and rating scale results with a DNA sample or a functional magnetic resonance imaging test to individualize treatment. Dr Thase responded that much of the variation in patient response to treatment comes from differences in drug metabolism rather than genetic differences in monoamine neurotransmission. However, genetic tests such as cheek swabs can indicate a patient’s potential drug metabolism as well. Neuroimaging tests may become worthwhile in the future, but they may be more cost-effective for treatment-resistant depression rather than routine baseline testing.
Mechanisms of Action of Antidepressant Classes
Next, Dr Thase reviewed the antidepressant medications historically by class (Table 2) and outlined their MOA, relative efficacy, and adverse effects.

Click figure to enlarge
Tricyclic Antidepressants
The TCAs were discovered serendipitously in the 1950s in an effort to develop safer antipsychotic medications.5 The TCAs currently approved by the FDA include imipramine, amitriptyline, desipramine, doxepin, nortriptyline, protriptyline, and trimipramine; an eighth TCA, clomipramine, is FDA-approved only for treatment of obsessive-compulsive disorder. Two other FDA-approved antidepressants, amoxapine and maprotiline, are sometimes grouped with the TCAs because of their close structural relationship. The TCAs nonselectively inhibit the reuptake of norepinephrine and, to a varying degree, serotonin, and have a range of effects on other receptor systems (eg, histaminergic, cholinergic).5 Although they were once considered the medication of first choice, today they are typically used for difficult-to-treat patients, and they appear later in treatment protocols due to their side effects and potential for death in overdose.5 Common adverse effects are dry mouth, blurred vision, constipation, tachycardia, sedation, weight gain, and hypotension.17 For patients over age 50 years, a cardiogram should be conducted before prescribing a TCA, and patients should be monitored for orthostatic hypotension if they report dizziness.18
The TCAs are predominately noradrenergic antidepressants, but clomipramine is a strongly serotonergic antidepressant with noradrenergic effects, making it beneficial for patients with severe depression or obsessive-compulsive disorder.5,19 Although not highly selective medications, nortriptyline and desipramine might be thought of as the best proven norepinephrine reuptake inhibitors and, with appropriate monitoring, are sometimes used in combinations with SSRIs and, on occasion, MAOIs.19
Monoamine Oxidase Inhibitors
MAOIs evolved as the counterpart to TCAs, although they were initially discovered when an antituberculosis drug improved depressive symptoms.5 If a TCA did not work, an MAOI was the next step because its multiple action (elevating serotonin, norepinephrine, and dopamine) made it a useful alternative for people who did not respond to the primarily noradrenergic effects of the TCAs.5
Nonselective MAOIs (that inhibit both MAO-A and MAO-B) include phenelzine, isocarboxazid, and tranylcypromine. Selegiline is sometimes described as a selective MAOI because it inhibits only MAO-B at low doses. However, oral selegiline, which is approved for treatment of Parkinson’s disease, is probably not an effective antidepressant at such low doses. Agents that inhibit MAO-A metabolize serotonin, norepinephrine, dopamine, and tyramine while MAO-B metabolizes phenylethylamine, dopamine, and tyramine.5 The inhibition of monoamine oxidase leads to a particular side effect that comes from the inability to break down tyramine in the gut. Thus, MAOIs may not be safely used unless the patient stays on a low tyramine diet, avoiding foods such as aged cheeses, soy products, tap beers, and certain wines.5 However, the transdermal patch delivery of selegiline is available as a treatment option without the dietary restrictions because it avoids enzyme inhibition in the gut and metabolism in the liver.18
Drugs that have strong serotonin effects are contraindicated in combination with MAOIs because of the risk of serotonin syndrome, a possible consequence of excess serotonin, which is characterized by symptoms including abdominal pain, diarrhea, sweating, lethargy, tremor, mental changes, and cardiovascular shock.18 The MAOIs are generally less sedating than the TCAs, and common adverse effects include hypotension, weight gain, sexual dysfunction, headaches, and insomnia.18
Despite their dietary restrictions and drug contraindications, MAOIs are very effective for a subset of patients who do not respond to other types of antidepressants. They are often considered after TCAs,18 but, for patients with atypical depression, an MAOI may be used before a TCA because the MAOIs have shown better efficacy for those patients than the TCAs.20 Atypical depression is characterized by mood reactivity and symptoms such as leaden paralysis, rejection sensitivity, and an increase in weight gain, appetite, or sleep.18
Norepinephrine-Dopamine Reuptake Inhibitor
The only FDA-approved norepinephrine-dopamine reuptake inhibitor (NDRI) for MDD is bupropion, which was first introduced in 1985, making it the oldest second-generation antidepressant. Because of a relatively high risk of seizures with the immediate-release, thrice-daily formulation, the drug was voluntarily withdrawn but then later returned with a maximum dose of 450 mg/d, which reduced the risk of seizures.5 Bupropion is a nonserotonergic agent with predominantly noradrenergic and dopaminergic reuptake inhibition, but these effects are relatively weak, so its metabolites’ potencies are thought to be related to its efficacy as an antidepressant.5 Its adverse effects include headache, dizziness, nausea, dry mouth, insomnia, and itching. Largely because it does not inhibit serotonin uptake, bupropion has no sexual side effects, making it a viable alternative for patients who had significant sexual dysfunction when taking one of the SSRIs.5
Selective Serotonin Reuptake Inhibitors
The SSRIs were first introduced with fluoxetine in the late 1980s,5 and they became the most widely used antidepressants in the United States and around the world by the 2000s.21,22 In most current treatment algorithms, the SSRIs are the first of the first-line treatments for patients with MDD.17,18 The SSRIs include citalopram, escitalopram, fluoxetine, paroxetine, sertraline, and fluvoxamine, although the latter medication is not approved by the FDA for treatment of MDD. As their name suggests, their MOA is selective and potent inhibition of serotonin reuptake.5 In addition to their well-tested antidepressant effects, these drugs are treatments of first choice for many anxiety disorders, which is useful for depressed patients with a concomitant or secondary anxiety disorder.5
Adverse effects associated with SSRIs include weight gain, dry mouth, nausea, headache, sleep problems, and sexual dysfunction. Despite their adverse effects, SSRIs are safer in overdose than TCAs23 and became the easier, safer alternatives to MAOIs and TCAs. Unlike the older drugs, SSRIs were widely prescribed for patients with milder depressions. The death by suicide rate is higher with the TCAs than with the SSRIs,23,24 although all antidepressant labels carry a warning about suicide risk, especially in youth. By virtue of their simplicity of action, SSRIs are fairly well tolerated, so they can be started at full therapeutic doses in most patients, whereas TCAs and MAOIs must be titrated gradually. Dr Thase noted, however, that inpatient studies have suggested that the TCA clomipramine is more effective than SSRIs for severe depression.25
Serotonin-Norepinephrine Reuptake Inhibitors
The SNRIs with FDA-approval for treating depression include venlafaxine, desvenlafaxine, duloxetine, and levomilnacipran. Again, as their name implies, the SNRIs block the reuptake of serotonin and norepinephrine. They are dual-mechanism drugs like TCAs, but with more selective actions like SSRIs to minimize side effects while maintaining therapeutic benefits.5 One review19 noted that duloxetine and venlafaxine may have less norepinephrine reuptake inhibition than some TCAs, making the TCAs more effective for severely depressed patients, although venlafaxine was still more effective than SSRIs in severe depression. The side effect profile of SNRIs is similar to that of SSRIs.
Although the SNRIs were developed as an effort to take the benefits of SSRIs and add antidepressant potency through a second MOA, whether the SNRIs have actually accomplished this goal has been a contentious topic,25 in part because determining when the norepinephrine contribution begins to become active is difficult with these agents. Evidence indicates that the SNRIs, on average, have a modest benefit over SSRIs, but the evidence is not equal across all of the SNRIs.25 As stated earlier, some depressive symptoms respond better to different antidepressant interventions (Case 2). For example, SNRIs may provide some advantage in patients who have significant pain complaints, and duloxetine is approved for several pain-related indications.26
Case 2
Ms M, a 36-year-old patient, is married with 2 children and works as a graphic artist and website designer. She was referred to a psychiatrist by her psychotherapist, who requested a reevaluation of her treatment and mood disorder, which the therapist believes is recurrent depression and possibly cyclothymic personality.
Ms M has a family history of suicide, depression, and alcohol and drug abuse. Her first depressive episode occurred when she was 16 years old, and she has had at least 4 subsequent periods of depression, including 2 postpartum episodes. Earlier episodes responded to psychotherapy and fluoxetine, but she relapsed each time. One episode occurred despite her taking fluoxetine 40 mg/d, although a hypothyroidism diagnosis and T4 treatment led to some quality-of-life improvement and stopped her weight gain. When sustained-release bupropion 150 mg twice daily was added to fluoxetine, Ms M seemed to improve but then experienced panic attacks, severe insomnia, and restlessness that led to discontinuation of this combination. She resumed psychotherapy and did quite well for several years, although she felt mildly depressed in the fall and winter.
Her latest episode was treated with extended-release venlafaxine, 75 mg/d, and Ms M felt “dramatically better” after only 2 weeks. However, her husband called to report that she was irritable and sleeping less, and her venlafaxine was discontinued at her next appointment.
At her follow-up visit 2 months later, Ms M presented with low energy and disinterest. Upon further questioning, the psychiatrist discovered that her mood switch on venlafaxine caused a hypomanic episode.
Ms M initially appeared to have recurrent depression. She relapsed even while taking adequate doses of antidepressants. However, she did show some identifying features of bipolar depression: she had an early age at onset of her first major depressive episode, cyclothymic temperament, more than 3 major depressive episodes, antidepressant-induced hypomania, postpartum depression, and seasonality. Her psychiatric history and current symptoms confirmed a pattern of depression mixed with hypomanic episodes. Her final diagnosis was bipolar II disorder.
Ms M was switched to a mood stabilizer and psychotherapy, which is a better treatment option for bipolar depression than antidepressant treatment.27 Some patients with bipolar forms of depression respond beautifully to antidepressants, whereas others develop rapid cycling or treatment-emergent affective shifts. It appears that dual reuptake inhibitors, including the TCAs and SNRIs, are more likely to cause such unwelcomed outcomes than SSRIs or bupropion. Clinicians must always assess for mania or hypomania in patients with depression to confirm or rule out bipolar disorder and plan to prophylax with a mood stabilizer when antidepressant medication is judged to be necessary.
Multimodal Antidepressants
The next class of antidepressants includes agents that modulate different receptors. Examples include mirtazapine, trazodone, nefazodone, vilazodone, and vortioxetine. With the exception of mirtazapine, these antidepressants build on serotonin reuptake inhibition by adding other mechanisms of action.
Mirtazapine, which is sometimes called a noradrenergic and specific serotonergic antidepressant (NaSSA), is not a reuptake inhibitor, but its serotonin and norepinephrine effects come from secondary mechanisms.5 A powerful antihistamine, mirtazapine is the most sedating of the newer antidepressants,28 and can cause weight gain and daytime drowsiness.5
Unlike mirtazapine, both trazodone and the structurally related drug nefazodone are weak serotonin reuptake inhibitors that also block serotonin-2A/2C receptors, classifying them as serotonin antagonist/reuptake inhibitors (SARIs).5 Trazodone and nefazodone both have sedating effects, and other common adverse events include nausea, dizziness, constipation, and blurred vision.29,30 Trazodone is commonly used off-label today as a sedative in low doses (25-100 mg), but at higher doses (150-600 mg) it can be an effective antidepressant.31 Once widely used because of a low risk of sexual side effects and a generally favorable effect on sleep, nefazodone is rarely used today because of the risk of hepatic toxicity.
The antidepressant vilazodone is a partial agonist at the serotonin-1A receptor in addition to inhibiting serotonin reuptake, so it has a second MOA. Its side effects include mild diarrhea and nausea; its side effect profile is distinguished by relatively low levels of weight gain and sexual dysfunction.32
Vortioxetine is a serotonin reuptake inhibitor that also has a number of secondary effects on other serotonin receptors, such as serotonin-1A/1B, serotonin-3, and serotonin-7,33 which may give it some advantage for patients with cognitive complaints.34 Generally, these antidepressants provide useful alternatives for patients who have not been able to tolerate SSRIs or SNRIs.5
Conclusion
Many patients with depression will have treatment-resistant symptoms that must be addressed. By considering whether patients’ specific symptoms indicate which monoamine pathway may be dysfunctioning, clinicians can try to select medications to target dopamine, norepinephrine, or serotonin in the brain. Different antidepressant classes are associated with specific changes in the 3 monoamine pathways. Altering these pathways brings therapeutic benefits and also adverse effects. Less selective action can lead to tolerability and safety issues, as with the TCAs and MAOIs, but these agents are effective for subsets of patients who do not respond to the generally safer SSRIs and SNRIs. Other antidepressants with selective receptor modulation can be effective to treat symptoms including insomnia, cognitive problems, and sexual dysfunction. Clinicians can target residual symptoms and treatment resistance by considering the MOA of their antidepressant options.
Drug names: aripiprazole (Abilify), bupropion (Aplenzin, Wellbutrin, and others), citalopram (Celexa and others), clomipramine (Anafranil and others), desipramine (Norpramin and others), desvenlafaxine (Pristiq, Khedezla, and others), doxepin (Zonalon, Silenor, and others), duloxetine (Cymbalta and others), escitalopram (Lexapro and others), fluoxetine (Prozac and others), fluvoxamine (Luvox and others), imipramine (Tofranil and others), isocarboxazid (Marplan), levomilnacipran (Fetzima), milnacipran (Savella), mirtazapine (Remeron and others), nortriptyline (Pamelor and others), paroxetine (Paxil, Pexeva, and others), phenelzine (Nardil), protriptyline (Vivactil and others), selegiline (EMSAM, Eldepryl, and others), sertraline (Zoloft and others), tranylcypromine (Parnate and others), trazodone (Oleptro and others), trimipramine (Surmontil and others), venlafaxine (Effexor and others), vilazodone (Viibryd), vortioxetine (Brintellix).
Disclosure of off-label usage: Dr Thase has determined that, to the best of his knowledge, clomipramine, fluvoxamine, and oral selegiline are not approved by the US Food and Drug Administration for the treatment of major depressive disorder.
Find more articles on this and other psychiatry and CNS topics:
The Journal of Clinical Psychiatry
The Primary Care Companion for CNS Disorders
References
1. Knoth RL, Bolge SC, Kim E, et al. Effect of inadequate response to treatment in patients with depression. Am J Manag Care. 2010;16(8):e188-e196. PubMed
2. Romera I, Pérez V, Ciudad A, et al. Residual symptoms and functioning in depression, does the type of residual symptom matter? a post-hoc analysis. BMC Psychiatry. 2013;13(1):51. PubMed doi:10.1186/1471-244X-13-51
3. Gartlehner G, Hansen RA, Thieda P, et al. Comparative Effectiveness of Second-Generation Antidepressants in the Pharmacologic Treatment of Adult Depression. Rockville, MD: Agency for Healthcare Research and Quality; 2007.
4. Nandi A, Beard JR, Galea S. Epidemiologic heterogeneity of common mood and anxiety disorders over the lifecourse in the general population: a systematic review. BMC Psychiatry. 2009;9(1):31. PubMed doi:10.1186/1471-244X-9-31
5. Stahl MS. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. 3rd ed. New York, NY: Cambridge University Press; 2008.
6. Sonawalla SB, Rosenbaum JF. Placebo response in depression. Dialogues Clin Neurosci. 2002;4(1):105-113. PubMed
7. Hamon M, Blier P. Monoamine neurocircuitry in depression and strategies for new treatments. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:54-63. PubMed doi:10.1016/j.pnpbp.2013.04.009
8. Prins J, Olivier B, Korte SM. Triple reuptake inhibitors for treating subtypes of major depressive disorder: the monoamine hypothesis revisited. Expert Opin Investig Drugs. 2011;20(8):1107-1130. PubMed doi:10.1517/13543784.2011.594039
9. Korte SM, Prins J, Krajnc AM, et al. The many different faces of major depression: it is time for personalized medicine. Eur J Pharmacol. 2015;753:88-104. PubMed doi:10.1016/j.ejphar.2014.11.045
10. Lane RM. Antidepressant drug development: focus on triple monoamine reuptake inhibition [published online ahead of print October 14, 2014]. J Psychopharmacol. 2014. PubMed doi:10.1177/0269881114553252
11. Hirschfeld RMA. History and evolution of the monoamine hypothesis of depression. J Clin Psychiatry. 2000;61(suppl 6):4-6. PubMed
12. Robinson DS. The role of dopamine and norepinephrine in depression. Prim Psychiatry. 2007;14(5):21-23.
13. Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry. 2007;64(3):327-337. PubMed doi:10.1001/archpsyc.64.3.327
14. Schatzberg AF. New approaches to managing psychotic depression. J Clin Psychiatry. 2003;64(suppl 1):19-23. PubMed
15. Drevets WC. Prefrontal cortical-amygdalar metabolism in major depression. Ann N Y Acad Sci. 1999;877:614-637. PubMed doi:10.1111/j.1749-6632.1999.tb09292.x
16. Monkul ES, Silva LA, Narayana S, et al. Abnormal resting state corticolimbic blood flow in depressed unmedicated patients with major depression: a (15)O-H(2)O PET study. Hum Brain Mapp. 2012;33(2):272-279. PubMed doi:10.1002/hbm.21212
17. Bauer M, Pfennig A, Severus E, et al, for the Task Force on Unipolar Depressive Disorders. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders, part 1: update 2013 on the acute and continuation treatment of unipolar depressive disorders. World J Biol Psychiatry. 2013;14(5):334-385. PubMed doi:10.3109/15622975.2013.804195
18. Gelenberg AJ, Freeman MP, Markowitz JC, et al. Practice Guideline for the Treatment of Patients With Major Depressive Disorder. Arlington, VA: American Psychiatric Association; 2010.
19. Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol. 2007;151(6):737-748. PubMed doi:10.1038/sj.bjp.0707253
20. Stewart JW, Thase ME. Treating DSM-IV depression with atypical features. J Clin Psychiatry. 2007;68(4):e10. PubMed doi:10.4088/JCP.0407e10
21. Nordqvist C. What are antidepressants? http://www.medicalnewstoday.com/articles/248320.php. Updated September 15, 2014. Accessed May 5, 2015.
22. Chee KY, Tripathi A, Avasthi A, et al. International study on antidepressant prescription pattern at 40 major psychiatric institutions and hospitals in Asia: a 10-year comparison study [published online ahead of print February 23, 2015]. Asia Pac Psychiatry. doi: 10.1111/appy.12176.
23. Hawton K, Bergen H, Simkin S, et al. Toxicity of antidepressants: rates of suicide relative to prescribing and non-fatal overdose. Br J Psychiatry. 2010;196(5):354-358. PubMed doi:10.1192/bjp.bp.109.070219
24. Gibbons RD, Hur K, Bhaumik DK, et al. The relationship between antidepressant medication use and rate of suicide. Arch Gen Psychiatry. 2005;62(2):165-172. PubMed doi:10.1001/archpsyc.62.2.165
25. Thase ME. Are SNRIs more effective than SSRIs? a review of the current state of the controversy. Psychopharmacol Bull. 2008;41(2):58-85. PubMed
26. Martinez JM, Katon W, Greist JH, et al. A pragmatic 12-week, randomized trial of duloxetine versus generic selective serotonin-reuptake inhibitors in the treatment of adult outpatients in a moderate-to-severe depressive episode. Int Clin Psychopharmacol. 2012;27(1):17-26. PubMed doi:10.1097/YIC.0b013e32834ce11b
27. Hirschfeld RMA. Guideline watch: practice guideline for the treatment of patients with bipolar disorder. 2nd ed. Published 2005. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/bpd-watch.pdf. Accessed May 5, 2015.
28. Thase ME. Depression and sleep: pathophysiology and treatment. Dialogues Clin Neurosci. 2006;8(2):217-226. PubMed
29. Oleptro (trazodone)[package insert]. Gaithersburg, MD: Angelini Pharma; 2010. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=88c45123-f475-4dd0-bbac-c261868924ef. Accessed May 5, 2015.
30. Nefazodone [package insert]. Sellersville, PA: Teva Pharmaceuticals; 2003. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=51ff7db5-aaf9-4c3c-86e6-958ebf16b60f. Accessed May 5, 2015.
31. Stahl SM. Mechanism of action of trazodone: a multifunctional drug. CNS Spectr. 2009;14(10):536-546. PubMed
32. Croft HA, Pomara N, Gommoll C, et al. Efficacy and safety of vilazodone in major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2014;75(11):e1291-e1298. PubMed doi:10.4088/JCP.14m08992
33. Brintellix (vortioxetine)[package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc; 2013. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=4b0700c9-b417-4c3a-b36f-de461e125bd3#nlm34084-4. Accessed May 5, 2015.
34. Leiser SC, Li Y, Pehrson AL, et al. Serotonergic regulation of prefrontal cortical circuitries involved in cognitive processing: a review of individual 5-HT receptor mechanisms and concerted effects of 5-HT receptors exemplified by the multimodal antidepressant vortioxetine [published online ahead of print March 6, 2015]. ACS Chem Neurosci. PubMed doi:10.1021/cn500340j


This PDF is free for all visitors!