Objective: To present a striking case of new-onset psychosis in a middle-aged woman subsequently diagnosed with behavioral variant frontotemporal dementia (bvFTD). To review the data regarding key red-flag features that may suggest a diagnosis of a neurodegenerative process, and specifically bvFTD, rather than a primary psychotic disorder. To examine the role of genetics, especially mutations of the microtubule-associated protein tau (MAPT) gene, in familial cases of frontotemporal dementia (FTD).
Data Sources: The pertinent literature was searched online (PubMed, Google Scholar) using the following search terms: frontotemporal dementia (FTD), Pick’s disease, behavioral variant FTD (bvFTD), psychosis, delusions, MAPT, and genetics. No date or language limit was applied.
Study Selection: The case report was generated through detailed assessment of clinical notes, imaging studies, and laboratory results. The brain autopsy was carried out and summarized by our neuropathology team. Previously published literature was selected for inclusion in the review section based on relevance to the topic.
Results: A neurodegenerative etiology for psychosis (and specifically bvFTD) should be suspected in patients with progressive deficits in executive function, language, or memory. Other key warning features include the presence of a strong family history of a late-life psychotic disorder (or institutional placement or suicide), loss of empathy, impaired recognition of facial expression, or the development of emotional blunting and apathy, abnormal movements, or seizures.
Conclusions: Neurodegenerative disease should be on the differential diagnosis for any patient presenting with new-onset psychosis and behavioral changes in mid to late adulthood. Should red-flag features be present, early referral to a clinic specializing in dementia is recommended for further evaluation. This case highlights that MAPT mutations can be associated with psychosis in FTD and should be considered in the genetic workup. Ongoing research into the cellular and neural circuit mechanisms of psychosis in neurodegenerative disease may shed light on pathologic processes underlying psychosis in primary psychiatric disorders.

CME Background
Articles are selected for credit designation based on an assessment of the educational needs of CME participants, with the purpose of providing readers with a curriculum of CME articles on a variety of topics throughout each volume. Activities are planned using a process that links identified needs with desired results.
To obtain credit, read the article, correctly answer the questions in the Posttest, and complete the Evaluation. A $10 processing fee will apply.
CME Objective
After studying this article, you should be able to:
‘ ¢Include neurodegenerative disease in the differential diagnosis for any patient presenting with new-onset psychosis and behavioral changes in mid-to-late adulthood
Accreditation Statement
The CME Institute of Physicians Postgraduate Press, Inc., is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Credit Designation
The CME Institute of Physicians Postgraduate Press, Inc., designates this journal-based CME activity for a maximum of 1 AMA PRA Category 1 Creditâ„¢. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Note: The American Academy of Physician Assistants (AAPA) accepts certificates of participation for educational activities certified for AMA PRA Category 1 Creditâ„¢ from organizations accredited by ACCME or a recognized state medical society. Physician assistants may receive a maximum of 1 hour of Category I credit for completing this program.
Release, Expiration, and Review Dates
This educational activity was published in March 2020 and is eligible for AMA PRA Category 1 Creditâ„¢ through April 30, 2022. The latest review of this material was March 2020.
Financial Disclosure
All individuals in a position to influence the content of this activity were asked to complete a statement regarding all relevant personal financial relationships between themselves or their spouse/partner and any commercial interest. The CME Institute has resolved any conflicts of interest that were identified. In the past year, Marlene P. Freeman, MD, Editor in Chief, has received research funding from JayMac and Sage; has been a member of the advisory boards for Otsuka, Alkermes, and Sunovion; has been a member of the Independent Data Safety and Monitoring Committee for Janssen; has been a member of the Steering Committee for Educational Activities for Medscape; and, as a Massachusetts General Hospital (MGH) employee, works with the MGH National Pregnancy Registry, which is sponsored by Teva, Alkermes, Otsuka, Actavis, and Sunovion, and works with the MGH Clinical Trials Network and Institute, which receives research funding from multiple pharmaceutical companies and the National Institute of Mental Health. No member of the CME Institute staff reported any relevant personal financial relationships. Faculty financial disclosure appears at the end of the article.
Objective: To present a striking case of new-onset psychosis in a middle-aged woman subsequently diagnosed with behavioral variant frontotemporal dementia (bvFTD). To review the data regarding key red-flag features that may suggest a diagnosis of a neurodegenerative process, and specifically bvFTD, rather than a primary psychotic disorder. To examine the role of genetics, especially mutations of the microtubule-associated protein tau (MAPT) gene, in familial cases of frontotemporal dementia (FTD).
Data Sources: The pertinent literature was searched online (PubMed, Google Scholar) using the following search terms: frontotemporal dementia (FTD), Pick’s disease, behavioral variant FTD (bvFTD), psychosis, delusions, MAPT, and genetics. No date or language limit was applied.
Study Selection: The case report was generated through detailed assessment of clinical notes, imaging studies, and laboratory results. The brain autopsy was carried out and summarized by our neuropathology team. Previously published literature was selected for inclusion in the review section based on relevance to the topic.
Results: A neurodegenerative etiology for psychosis (and specifically bvFTD) should be suspected in patients with progressive deficits in executive function, language, or memory. Other key warning features include the presence of a strong family history of a late-life psychotic disorder (or institutional placement or suicide), loss of empathy, impaired recognition of facial expression, or the development of emotional blunting and apathy, abnormal movements, or seizures.
Conclusions: Neurodegenerative disease should be on the differential diagnosis for any patient presenting with new-onset psychosis and behavioral changes in mid to late adulthood. Should red-flag features be present, early referral to a clinic specializing in dementia is recommended for further evaluation. This case highlights that MAPT mutations can be associated with psychosis in FTD and should be considered in the genetic workup. Ongoing research into the cellular and neural circuit mechanisms of psychosis in neurodegenerative disease may shed light on pathologic processes underlying psychosis in primary psychiatric disorders.
J Clin Psychiatry 2020;81(2):19r13027
To cite: Ferenczi EA, Erkkinen MG, Feany MB, et al. New-onset delusions heralding an underlying neurodegenerative condition: a case report and review of the literature. J Clin Psychiatry. 2020;81(2):19r13027.
To share: https://doi.org/10.4088/JCP.19r13027
© Copyright 2020 Physicians Postgraduate Press, Inc.
aDepartment of Neurology, Brigham and Women’s Hospital, Boston, Massachusetts
bDepartment of Neurology, Massachusetts General Hospital, Boston, Massachusetts
cCenter for Brain/Mind Medicine, Brigham and Women’s Hospital, Boston, Massachusetts
dDepartment of Pathology, Brigham and Women’s Hospital, Boston Massachusetts
eDepartment of Psychiatry, Brigham and Women’s Hospital, Boston, Massachusetts
*Corresponding author: Emily A. Ferenczi, BM, BCh, PhD, Department of Neurology, Massachusetts General Hospital, 55 Fruit St, Boston, MA 02114 ([email protected]).
Most primary psychotic disorders are diagnosed in early adulthood; however, a notable minority of individuals are diagnosed after the age of 40 years.1 These individuals pose a diagnostic challenge, as the overlap with neurodegenerative causes of psychosis becomes increasingly prevalent with advancing age. A neurodegenerative etiology for psychosis should be suspected in any patient with progressive deficits in executive function, language, or memory. Other red-flag features may include new-onset seizures, abnormal movements, or a prominent family history of a dementing or progressive psychotic illness in mid to late adulthood (summary provided in Table 1). In such cases, a number of diagnostic tests are recommended (overview provided in Table 2).2,3 Should a neurodegenerative etiology be suspected, prompt referral to a clinic specializing in dementia is important to aid early diagnosis, ensure optimal care for the patient, and provide counseling and guidance for family or guardians. In this review, we focus specifically on the clinical features that may suggest a diagnosis of behavioral variant frontotemporal dementia (bvFTD). We emphasize that the MAPT mutation can be a cause of late-onset psychosis, as exemplified by the case of Ms A, which follows.
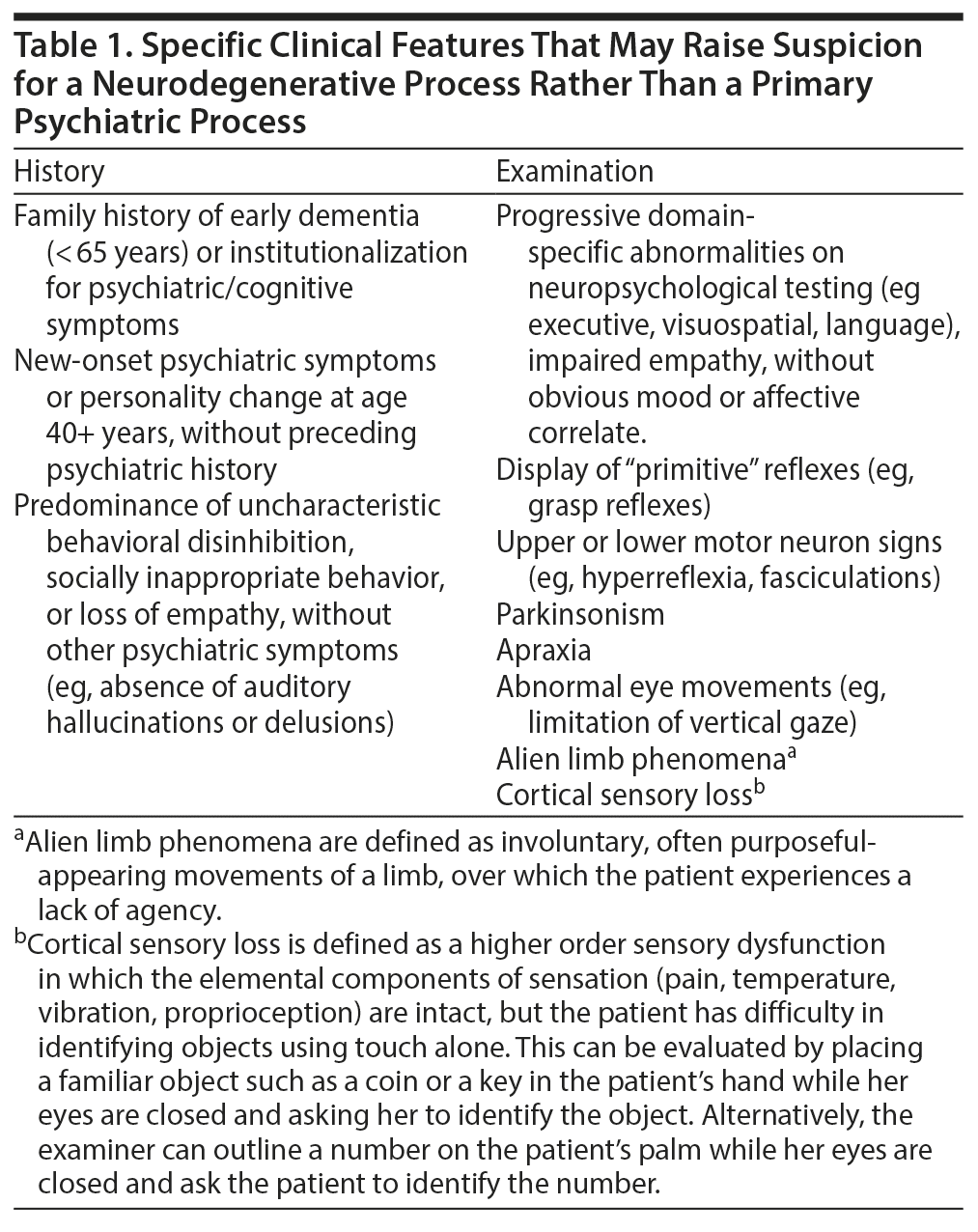
Click figure to enlarge
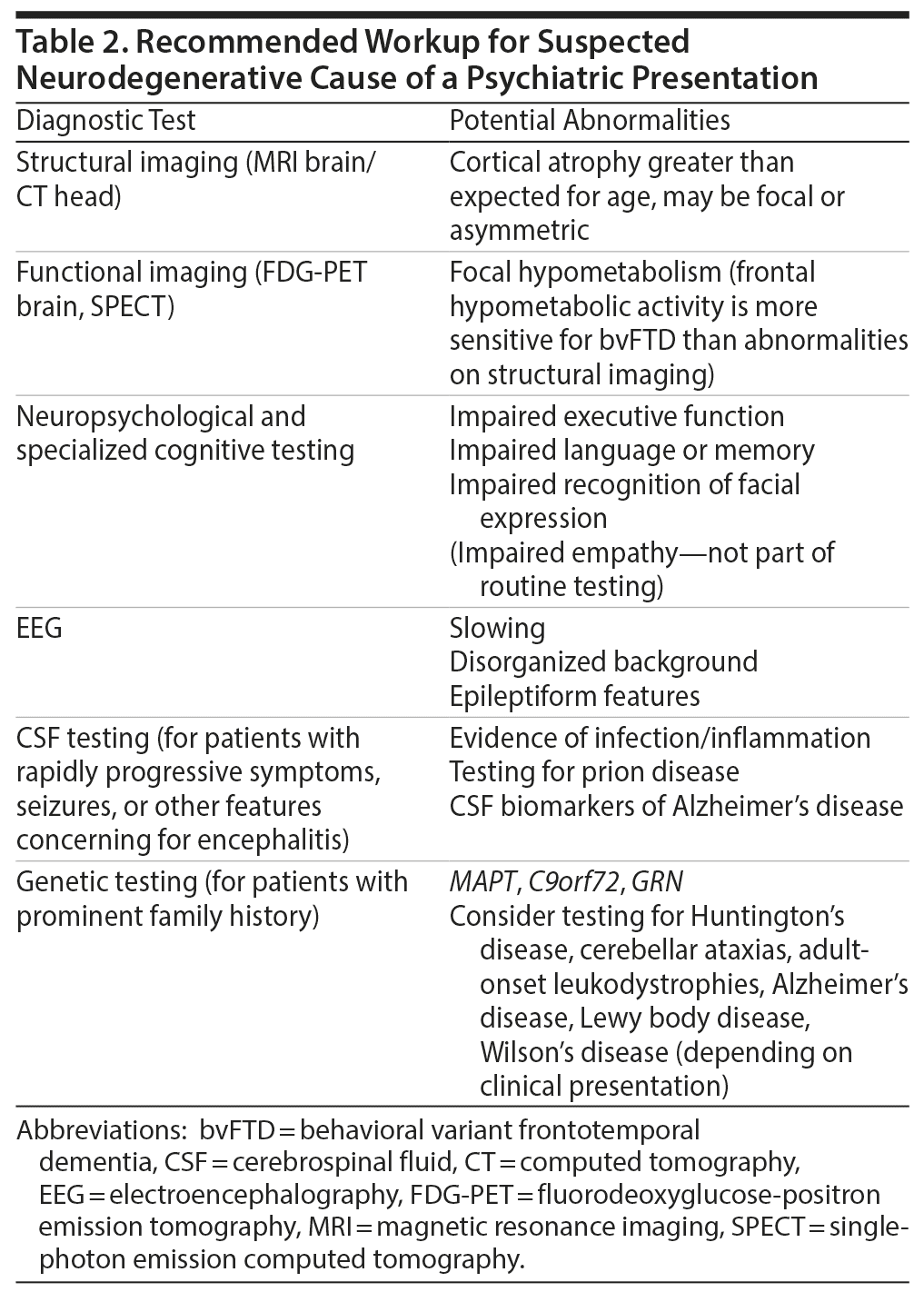
Click figure to enlarge
CASE REPORT
Ms A, a 39-year-old woman, was brought for an evaluation after 18 months of changes in personality, behavior, and judgment. She had separated from her husband, and the Department of Social Services had taken over custody of her children due to concern for neglect. She became homeless, although she strongly asserted to be living in an apartment she had bought. She also claimed to have $49,000 in a local bank, which was not true. She declared that her estranged father had died and that his ashes had been sent to her, although relatives denied this. There was no personal history of psychiatric illness or major medical illness. Her mother and maternal grandfather both experienced progressive psychiatric symptoms with onset in their 40s. Her mother died aged 51 (cause of death not available).
Troubled by the dramatic change in Ms A’s behavior, a close friend reached out to a neurologist at our hospital (K.R.D.) to request an evaluation. At her first neurology visit, Ms A exhibited labile affect. Her language was fluent and prosodic, and she could generate 14 words beginning with the letter S. Digit span was 7 digits forward, 5 backward. Her clock drawing was appropriate and there were no frontal release signs. Other aspects of the neurologic examination findings were normal.
Two months after the first clinic visit, Ms A was arrested and taken to jail after stealing a van from her ex-husband‘ s house. She was sent to a local emergency department for evaluation, where she claimed that the policeman who arrested her “made love to her and made her breakfast in the morning at the jail.” At a neurology clinic visit several weeks later, she scored 30/30 on the Mini-Mental State Examination, but her thoughts were delusional and she was deemed unsafe to manage her own affairs. She was admitted to a psychiatric hospital, where neuropsychological testing demonstrated deficits in executive function but intact memory. The results of structural brain imaging were not available. A fluorodeoxyglucose positron emission tomography (FDG-PET) brain scan revealed decreased metabolism in the frontal and temporal poles and caudate heads bilaterally (Figure 1). Genetic testing (Mayo Clinic Laboratories) detected a DNA sequence change in the microtubule-associated protein tau (MAPT) gene at exon 10 (1907C > T) corresponding to an amino acid change of proline 636 to leucine (P301L). This result was consistent with frontotemporal lobar degeneration due to a tauopathy linked to a mutation in the MAPT gene.
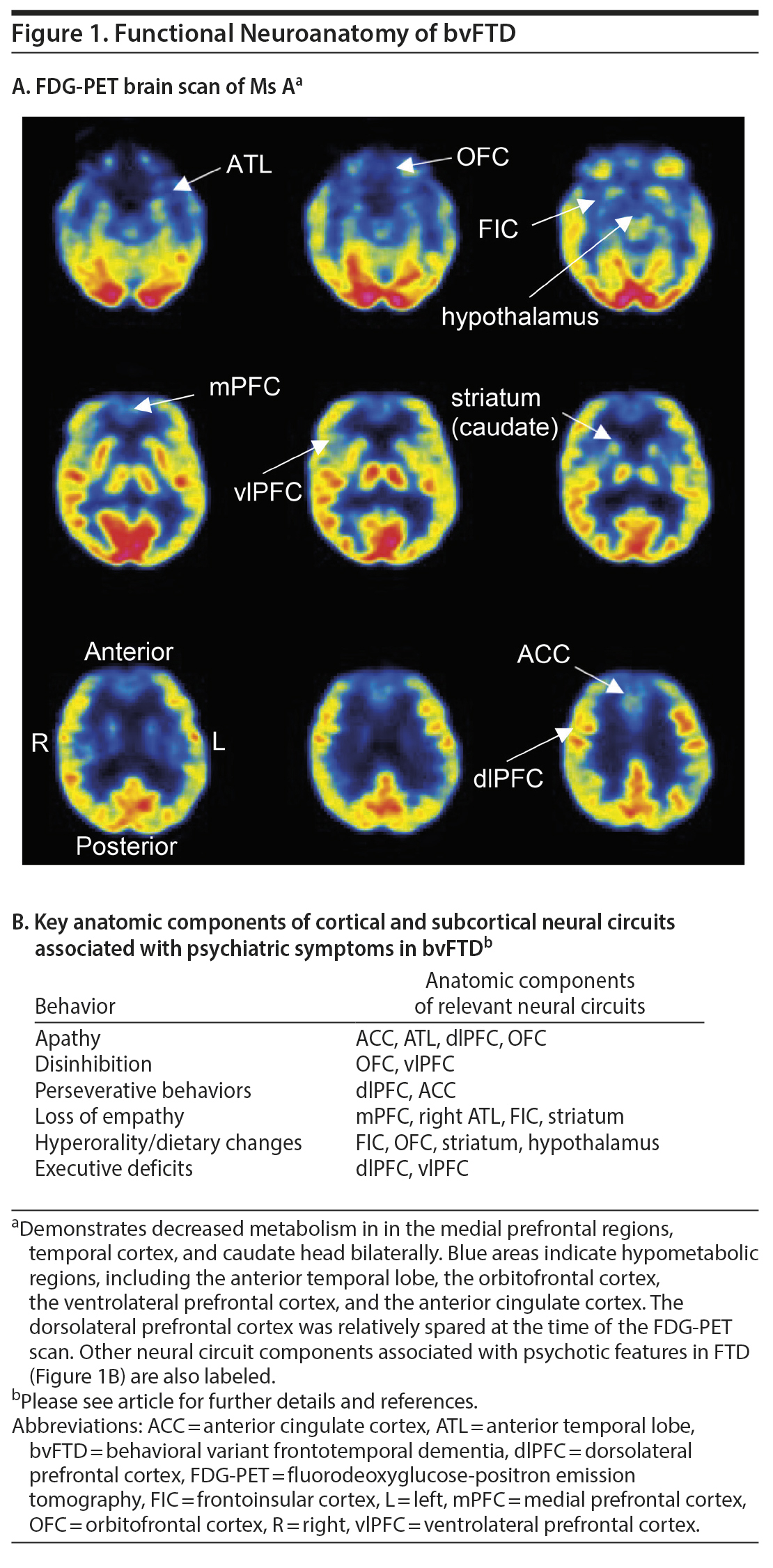
Click figure to enlarge
During subsequent years, Ms A’s symptoms progressed, and she required full-time supervision in a locked unit in a nursing home. At 5 years from presentation, she exhibited increasing agitation, repetitive behaviors, and inappropriate actions such as eating her own vomit and getting into other patients’ beds. She produced no spontaneous verbal output but was able to sing songs from memory. At 10 years, she could not sit or lie down for more than a few minutes. She put any object she encountered into her mouth. She showed no recognition of friends or family. She died at age 49, 10.5 years after her initial presentation, from presumed aspiration.
An autopsy examination of the brain revealed significant frontotemporal atrophy. The microscopic pathology was consistent with frontotemporal lobar degeneration (described in detail in Figure 2).
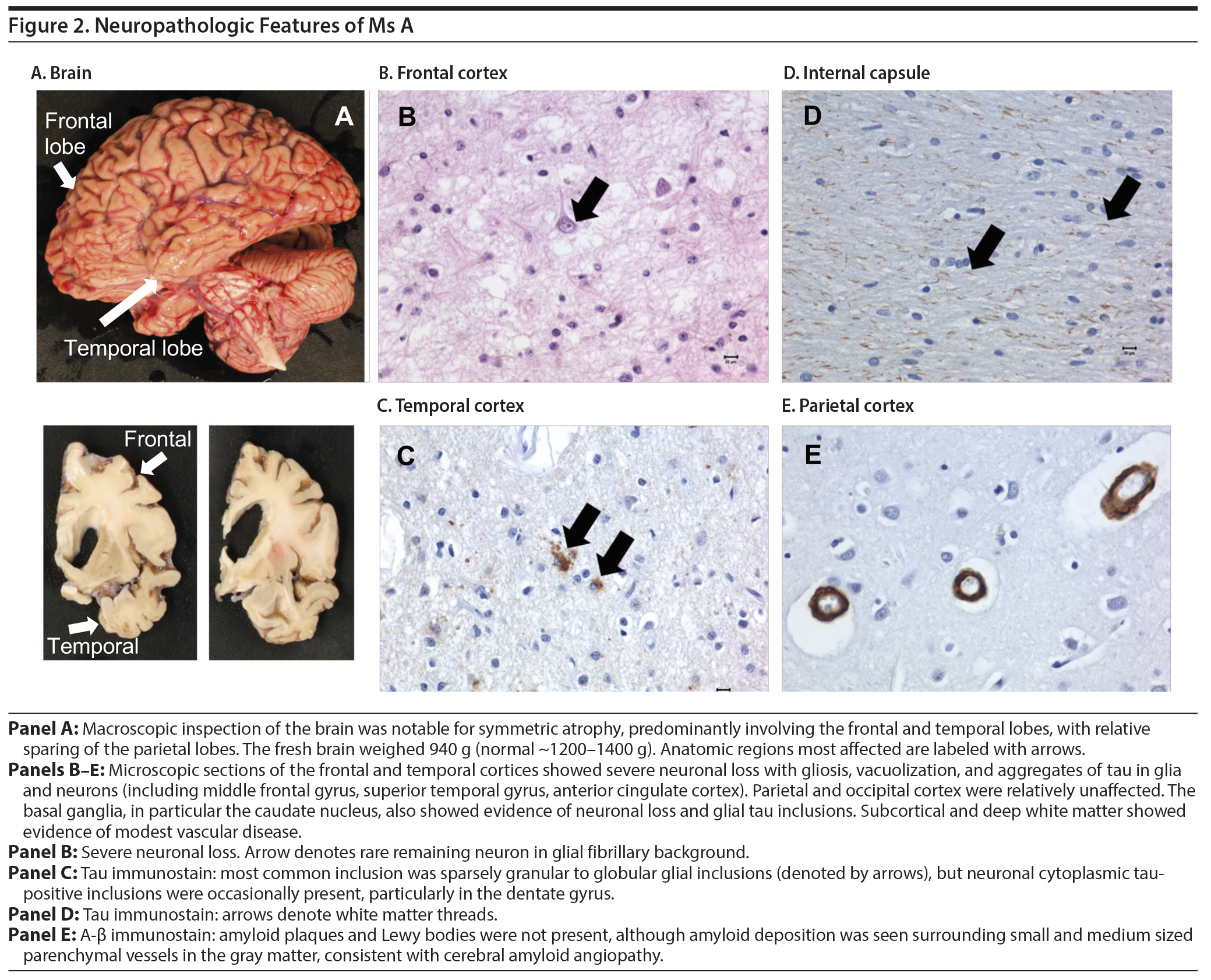
Click figure to enlarge
DISCUSSION
Behavioral Variant Frontotemporal Dementia as a Cause of New-Onset Psychosis
In 1892, Czech psychiatrist Arnold Pick described a 71-year-old man with “progressive loss of language and a failing mind,” with asymmetric cortical atrophy at autopsy. In the early 1920s, Alzheimer identified the characteristic intracellular inclusions associated with this atrophy, labeling them Pick bodies, and shortly thereafter the condition was named “Pick’s disease.”4 It has since become apparent that the original Pick’s disease is one subtype of frontotemporal lobar degeneration (FTLD), characterized by the aggregation of insoluble protein in neurons and glial cells. FTLD neuropathology is associated with a spectrum of clinical phenotypes that are collectively known as frontotemporal dementia (FTD). The expressed clinical syndrome depends primarily on the neuroanatomic location of pathology. The current classification subdivides FTD into 3 subtypes: bvFTD, nonfluent/agrammatic variant primary progressive aphasia, and semantic variant primary progressive aphasia. bvFTD is the most common variant in the United States and Europe, accounting for 50%-60% of FTD cases in these regions.5,6
- Identifying cases of new-onset psychosis later in life due to neurodegenerative disease such as frontotemporal dementia can be challenging.
- A neurodegenerative etiology should be suspected in patients with a strong family history, progressive decline in executive functioning, loss of empathy, or abnormal movements, which should lead to a dementia specialty clinic referral.
- MAPT gene mutations can be associated with psychosis in frontotemporal dementia and should therefore be part of the genetic workup.
Patients with bvFTD typically present with a preponderance of psychiatric and behavioral symptoms.7-10 Usually, there is relative sparing of sensory-motor functioning and certain aspects of cognition (such as memory and visuospatial skills). As a result, patients often present to psychiatric services before their neurologic diagnosis is made. One retrospective study revealed that 52% of patients diagnosed with bvFTD had previously received a diagnosis of a primary psychiatric disorder.11 This is not surprising since the estimated prevalence of FTD is approximately 2 orders of magnitude lower than that of primary psychotic disorders; 15-22 per 100,000 for FTD6 vs ~4 per 1,000 for primary psychosis.12 However, such misdiagnoses can have the negative effect of isolating patients from family and friends13 and may increase the risk of institutionalization for criminal behavior either in jail or in psychiatric institutions14 (more common in FTD than in Alzheimer’s disease15).
In bvFTD, neuropsychiatric symptoms are present in the majority of patients and often reflect the specific cortical and subcortical neural circuits affected by the pathology (Figure 1B). Negative symptoms, such as apathy, inertia, and emotional blunting, tend to be encountered frequently and early in the disease course16 and have been linked to dysfunction in the anterior cingulate cortex and anterior temporal lobe (ATL).5 Social behavior problems, difficulty with emotional processing, and loss of empathy are also highly characteristic of bvFTD and are associated with right-sided brain abnormalities,17 particularly in the ATL, frontoinsular regions, anterior rostral medial frontal regions, and striatum.5,18 These brain regions, as well as others, including the temporoparietal junction, superior temporal sulcus, and amygdala, have been associated with theory of mind (awareness of another person’s mental state) and affective perspective-taking (ability to infer another person’s emotions), which are critical for empathy.19 Other prominent deficits, such as disinhibition and compulsions, typically occur later in the bvFTD disease course and are thought to reflect the anatomic progression of pathology into the orbitofrontal and frontoinsular cortex.5,17 Changes in eating behavior in FTD, another late feature, appear to involve changes in the right ventral anterior insula, striatum, and orbitofrontal cortex,20 as well as the hypothalamus.21 The brain regions associated with psychosis may vary for different FTD genetic mutations. For example, a quantitative imaging study found that psychotic symptoms in FTD are correlated with gray matter atrophy in select subcortical and cortical circuits including anterior insular, left thalamus, and cerebellum in GRN mutation carriers; left frontal lobe in C9orf72 mutation carriers; and temporal lobe in MAPT mutation carriers.22
Although no single clinical feature is pathognomonic for FTD psychosis rather than a primary psychotic disorder, certain abnormalities of thought processing, such as difficulty with abstract reasoning, stereotypical thinking, and repetitive or ritualized behaviors, are more commonly associated with FTD than primary psychotic disorders,16 although the precise neural substrate for these symptoms remains unknown. A smaller proportion of FTD patients (~20%-30%) exhibit positive psychotic symptoms such as paranoid delusions and hallucinations,16,23 and these are seen at higher frequency (40%-60%) in patients with specific genetic FTD variants, such as the C9orf72 mutation.24-26 The content of psychotic thoughts can be variable in FTD. For example, a striking feature of Ms A’s delusions was their positive or even fantastical quality, which has been noted in other case reports of FTD.27 However, given a lack of systematic study comparing clinical data on the phenomenological differences between FTD psychosis and idiopathic primary psychosis, the content and quality of delusions may not represent a reliable distinguishing feature. In several case series, the reported delusions varied across patients, from paranoid or persecutory to grandiose (involving famous people) or somatic (eg, body part distortion or parasitosis).28,29 It is also not always clear whether such disordered thoughts are true delusions (fixed beliefs) versus confabulations (more fleeting in nature, as may have been the case for Ms A). It is our clinical impression that the emotional impact of delusions may sometimes help to distinguish bvFTD from a primary psychiatric process. We have found that in FTD, the psychosis tends to be less emotionally salient or distressing to the patient and less associated with feelings of anxiety, guilt, and tension, perhaps in the setting of the global loss of emotional awareness that is characteristic of the condition.16
The presence of a strong family history of a late-life psychotic disorder, early-onset dementia, or placement in an institution should raise suspicion for an underlying neurodegenerative condition. An estimated 20%-40% of patients with FTD have a positive family history, and this follows an autosomal dominant pattern in the majority of cases.30 It is important to specifically ask whether any family member was institutionalized or died young with psychiatric or neurologic symptoms or from suicide, and it may be necessary to probe deeply as these family members may be forgotten or rarely talked about, due to associated stigma. Family history is also an important risk factor in primary psychotic disorders (~10%-30% of schizophrenia31); however, progression to death over several years is less common.
The temporal progression of disease will help to differentiate neurodegenerative from primary psychiatric disorders, although predicting the course of illness is often most difficult during the early stages of the illness when diagnostic uncertainty remains high. Over a timeframe of months to years, close attention should be paid to the development of neurologic signs suggestive of frontal network disease or motor neuron or basal ganglia dysfunction, as well as domain-specific impairments on neuropsychologic testing. Approximately 12.5% of bvFTD patients develop neurologic signs consistent with motor neuron pathology (eg, amyotrophic lateral sclerosis), including upper and lower motor neuron signs, dysarthria, and pseudobulbar affect. In 20% of patients, a parkinsonian syndrome may develop, which sometimes includes features of corticobasal syndrome (CBS; can be associated with asymmetric motor signs and alien limb phenomena—involuntary, often purposeful-appearing movements of a limb, over which the patient experiences a lack of agency) or progressive supranuclear palsy (PSP, associated with loss of balance and impaired voluntary eye movements).14 In some cases, the temporal order of these symptoms is reversed, with motor symptoms being most salient initially or concurrent with behavioral changes.
Full neuropsychological testing may reveal progressive domain-specific impairments, particularly in executive function (for example, on the Wisconsin Card Sorting task, the Stroop test, the Controlled Oral Word Association Test, and the Trail Making Test—Parts A and B). Unfortunately, many patients with primary psychiatric disease also show deficits in executive function. Therefore, this pattern of performance is not entirely specific for FTD. In addition, the degree of behavior and personality changes in bvFTD is often out of proportion to the executive dysfunction captured by neuropsychological testing. More specific deficits may include vulnerabilities in semantic knowledge, less likely to be affected in psychiatric disease, as well as deficits in emotional and social cognition. Recognition of facial expression is often profoundly impaired in FTD and most prominent for negative emotions such as fear and disgust.5 Patients with FTD also have significantly reduced empathy scores compared to normal controls, as reported by caregivers.18 These reflect a diminished capacity to imagine the perspective of another (cognitive empathy) and a reduced emotional response to the perception of another individual’s emotional state (emotional empathy). Patients with schizophrenia may also have reduced empathy compared to patients with bipolar disorder or major depression and healthy controls32; however, this deficit is generally felt to be less salient compared to patients with FTD.33 However, it is important to note that tests of facial recognition and empathy are not part of routine neuropsychological testing, and although new tools for emotional and social assessment are being developed (eg, EMOTICOM test battery),34 these have not yet been robustly validated across different patient populations and cultures.
Overall, it is important to note that although psychotic symptoms can be observed in FTD, they rarely occur in isolation and are usually accompanied by marked and progressive changes in behavior and personality, including loss of insight, executive function, empathy, and disordered eating. A useful bedside clinical tool (18 question checklist) has recently been developed by Ducharme et al,35 for which a score of 11 or greater is strongly suggestive of bvFTD (specificity 93.9%, sensitivity 71.1%), whereas a score of 8 or less is strongly suggestive of a primary psychiatric disorder (specificity 91.3%, sensitivity 77.3%). The checklist incorporates key distinguishing features including prior history of psychiatric symptoms, family history, degree of emotional distress or awareness of current situation, duration of symptoms, presence of stereotypical behaviors, changes in food preferences, and presence or absence of abnormalities on elemental neurologic examination. Ms A would have scored a total of 14 points at initial presentation to the neurology clinic (13 “no” answers in Part A + 1 “yes” answer in Part B, for the possible history of Pick’s disease in the patient’s mother). Five years into the disease course, Ms A’s score would have increased to 16, to take into account her development of stereotypical behaviors and changes in food preference.
MAPT Mutation in bvFTD
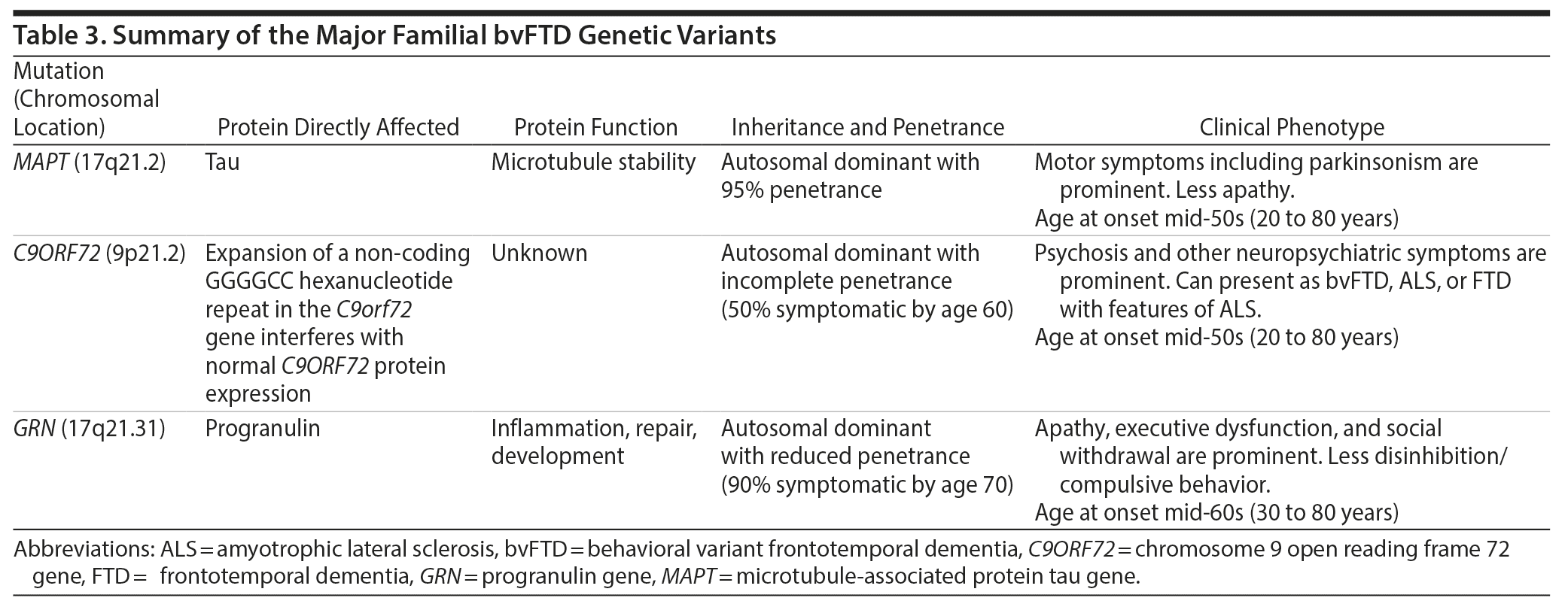
A key feature in Ms A’s history was her family history of an early-onset, progressive neuropsychiatric disorder in conjunction with a genetic mutation in the MAPT gene. Through investigations of such familial cases, several gene mutations have now been linked to FTD, and increased understanding of these mutations and their downstream effects on protein and neural circuit function is helping to reveal mechanisms underlying the neurodegenerative pathology. The most common mutations are found on 3 genes: MAPT, C9orf72, and GRN (progranulin). In this review, we focus on the MAPT mutations. Other mutations are discussed in detail elsewhere5,14,36 and are summarized in Table 3.
Click figure to enlarge
The MAPT gene mutation was first identified in a family with autosomal dominant inheritance, in which the mutation was linked to chromosome 17q21.2,37 later identified as the MAPT gene.38-40 Patients with this mutation tend to present earlier than those with C9orf72 or GRN mutations (50% with age at onset < 50 years), but the range of onset age is broad.25,41 The average disease duration is 8 years, with a younger average age at death (62 years) than patients with C9orf72 or GRN mutations; however, this can also range widely.25 In terms of clinical presentation, patients with the MAPT mutation may have less apathy but more prominent motor symptoms (most commonly levodopa-unresponsive parkinsonism) than other genetic variants. Another distinguishing feature is impaired language, particularly errors in word comprehension and object naming.25 However, the phenotype even within a single family carrying a single MAPT mutation can vary, with some members carrying a clinical diagnosis of bvFTD and other members corticobasal syndrome.
An important point arising from the case of Ms A is that patients with a MAPT mutation can present with psychosis, a symptom that has been more commonly associated with the C9orf72 and, to a lesser extent, GRN mutations.25 Thus, MAPT should be included, along with C9orf72 and GRN, as part of any genetic testing panel for patients with suspected FTD psychosis. Structural brain imaging characteristics may help point toward a MAPT mutation, which may show more prominent temporal lobe than frontal lobe atrophy on MRI than do other genetic mutations.25
Under physiological conditions, the tau protein plays an important role in microtubule stabilization. Different MAPT mutations influence the tendency of tau to become hyperphosphorylated and form insoluble neurofibrillary tangles, thus leading to cytoskeletal instability and neurotoxicity to differing extents.36,42 The most common mutation (P301 L), found in Ms A, is a missense point mutation in exon 10, which reduces the ability of the tau protein to interact with microtubules.43,44 However, despite the knowledge afforded by these familial mutations, the amount and type of tau pathology do not yet appear to predict a single clinical syndrome with high accuracy (eg, bvFTD vs CBS vs PSP), suggesting that additional factors may influence the clinical expression of disease.
Recommendations for the Psychiatrist
For patients with any red-flag features, including new-onset psychosis over the age of 40, a symptom course that is refractory to treatment or progressive over time, and/or a strong family history of neurodegenerative disease, we recommend that an elemental neurologic examination be performed. In agreement with recent recommendations regarding the importance of the physical examination in psychiatry,45,46 we suggest that even a brief neurologic examination, including evaluation for abnormal eye movements, frontal release signs, parkinsonism, and cerebellar and upper or lower motor neuron signs, could provide important early diagnostic information. A 3-minute neurologic examination for the psychiatrist is described by Garden.47 In addition, structural brain imaging with MRI or CT should be obtained to look for focal atrophy and exclude other structural abnormalities that may account for the patient’s presentation. If diagnostic uncertainty remains, we recommend prompt referral to a clinic specializing in dementia, where a detailed cognitive examination, further neuropsychiatric testing, and functional brain imaging can be pursued, as indicated. Functional brain imaging, for example with FDG-PET, provides additional diagnostic sensitivity for bvFTD (90%); however, results from this neuroimaging technique are associated with lower specificity (68%) because patients with primary psychiatric diagnoses can also exhibit regional hypometabolism, predominantly in the bilateral frontal and temporal lobes.48 However, in our experience, it would be exceedingly rare for a patient with a primary psychiatric diagnosis to exhibit the profound hypometabolic activity observed on Ms A’s scan.
CONCLUSIONS
Frontotemporal dementia should be on the differential diagnosis for any patient presenting with new-onset psychosis and behavioral changes in mid to late adulthood. A detailed neurologic and psychiatric family history should be obtained, and specific features of aberrant behaviors elicited, such as progressive personality change, uncharacteristic socially inappropriate behavior, or loss of empathy. A brief neurologic examination (including gait, eye movements, cerebellar examination, muscle tone, and reflexes) should be performed and structural brain imaging obtained. Should concerning features in the history, examination, or imaging be present, early referral to a clinic specializing in neurodegenerative disease/dementia is recommended. This report has several limitations. It includes only a single case that was retrospectively constructed, which may restrict its generalizability across the entire FTD spectrum. However, this case serves to highlight that the MAPT mutation can be associated with psychosis in FTD, and investigation of this mutation may contribute to the understanding of the neuroanatomical circuits and cellular mechanisms underlying psychiatric symptoms in neurodegenerative disease.
Submitted: July 31, 2019; accepted January 8, 2020.
Published online: March 31, 2020.
Disclosure of off-label usage: The authors have determined that, to the best of their knowledge, no investigational information about pharmaceutical agents or device therapies that is outside US Food and Drug Administration-approved labeling has been presented in this activity.
Financial disclosure: Drs Ferenczi, Erkkinen, Feany, Fogel, and Daffner have no personal affiliations or financial relationships with any commercial interest to disclose relative to the article.
Funding/support: No external funding sources were required for this work.
Acknowledgments: The authors thank the patient’s guardian for granting permission to publish this case and for being such a caring friend during the patient’s life.
Patient consent: The patient’s guardian provided consent to publish this case, and information has been de-identified to protect anonymity.
REFERENCES
1.Maglione JE, Thomas SE, Jeste DV. Late-onset schizophrenia: do recent studies support categorizing LOS as a subtype of schizophrenia? Curr Opin Psychiatry. 2014;27(3):173-178. PubMed CrossRef
2.Geschwind MD. Rapidly progressive dementia. Continuum (Minneap Minn). 2016;22(2 dementia):510-537. PubMed
3.Mendez MF, Shapira JS, McMurtray A, et al. Accuracy of the clinical evaluation for frontotemporal dementia. Arch Neurol. 2007;64(6):830-835. PubMed CrossRef
4.Pearce JMS. Pick’s disease. J Neurol Neurosurg Psychiatry. 2003;74(2):169. PubMed CrossRef
5.Lanata SC, Miller BL. The behavioural variant frontotemporal dementia (bvFTD) syndrome in psychiatry. J Neurol Neurosurg Psychiatry. 2016;87(5):501-511. PubMed CrossRef
6.Onyike CU, Diehl-Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry. 2013;25(2):130-137. PubMed CrossRef
7.Waddington JL, Youssef HA, Farrell MA, et al. Initial “schizophrenia-like” psychosis in Pick’s disease: case study with neuroimaging and neuropathology, and implications for frontotemporal dysfunction in schizophrenia. Schizophr Res. 1995;18(1):79-82. PubMed CrossRef
8.Snowden JS, Neary D. Neuropsychiatric aspects of frontotemporal dementias. Curr Psychiatry Rep. 1999;1(1):93-98. PubMed CrossRef
9.Mendez MF, Shapira JS, Woods RJ, et al. Psychotic symptoms in frontotemporal dementia: prevalence and review. Dement Geriatr Cogn Disord. 2008;25(3):206-211. PubMed CrossRef
10.Mendez MF, Lauterbach EC, Sampson SM; ANPA Committee on Research. An evidence-based review of the psychopathology of frontotemporal dementia: a report of the ANPA Committee on Research. J Neuropsychiatry Clin Neurosci. 2008;20(2):130-149. PubMed CrossRef
11.Woolley JD, Khan BK, Murthy NK, et al. The diagnostic challenge of psychiatric symptoms in neurodegenerative disease: rates of and risk factors for prior psychiatric diagnosis in patients with early neurodegenerative disease. J Clin Psychiatry. 2011;72(2):126-133. PubMed CrossRef
12.Moreno-Küstner B, Mart×n C, Pastor L. Prevalence of psychotic disorders and its association with methodological issues: a systematic review and meta-analyses. PLoS One. 2018;13(4):e0195687. PubMed CrossRef
13.Chemali Z, Withall A, Daffner KR. The plight of caring for young patients with frontotemporal dementia. Am J Alzheimers Dis Other Demen. 2010;25(2):109-115. PubMed CrossRef
14.Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672-1682. PubMed CrossRef
15.Liljegren M, Naasan G, Temlett J, et al. Criminal behavior in frontotemporal dementia and Alzheimer disease. JAMA Neurol. 2015;72(3):295-300. PubMed CrossRef
16.Gossink FT, Vijverberg EG, Krudop W, et al. Psychosis in behavioral variant frontotemporal dementia. Neuropsychiatr Dis Treat. 2017;13:1099-1106. PubMed CrossRef
17.Seeley WW, Bauer AM, Miller BL, et al. The natural history of temporal variant frontotemporal dementia. Neurology. 2005;64(8):1384-1390. PubMed CrossRef
18.Rankin KP, Gorno-Tempini ML, Allison SC, et al. Structural anatomy of empathy in neurodegenerative disease. Brain. 2006;129(pt 11):2945-2956. PubMed CrossRef
19.Healey ML, Grossman M. Cognitive and affective perspective-taking: evidence for shared and dissociable anatomical substrates. Front Neurol. 2018;9:491. PubMed CrossRef
20.Woolley JD, Gorno-Tempini M-L, Seeley WW, et al. Binge eating is associated with right orbitofrontal-insular-striatal atrophy in frontotemporal dementia. Neurology. 2007;69(14):1424-1433. PubMed CrossRef
21.Piguet O, Petersén A, Yin Ka Lam B, et al. Eating and hypothalamus changes in behavioral-variant frontotemporal dementia. Ann Neurol. 2011;69(2):312-319. PubMed CrossRef
22.Sellami L, Bocchetta M, Masellis M, et al; Genetic FTD Initiative, GENFI. Distinct neuroanatomical correlates of neuropsychiatric symptoms in the three main forms of genetic frontotemporal dementia in the GENFI cohort. J Alzheimers Dis. 2018;65(1):147-163. PubMed
23.Landqvist Waldö M, Gustafson L, Passant U, et al. Psychotic symptoms in frontotemporal dementia: a diagnostic dilemma? Int Psychogeriatr. 2015;27(4):531-539. PubMed CrossRef
24.Devenney EM, Landin-Romero R, Irish M, et al. The neural correlates and clinical characteristics of psychosis in the frontotemporal dementia continuum and the C9orf72 expansion. Neuroimage Clin. 2016;13:439-445. PubMed CrossRef
25.Snowden JS, Adams J, Harris J, et al. Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(7-8):497-505. PubMed CrossRef
26.Shinagawa S, Nakajima S, Plitman E, et al. Psychosis in frontotemporal dementia. J Alzheimers Dis. 2014;42(2):485-499. PubMed CrossRef
27.Kremen SA, Solis OE, Shapira JS, et al. “Fantastic thinking” in pathologically proven Pick disease. Cogn Behav Neurol. 2010;23(2):130-134. PubMed CrossRef
28.Mendez MF, Fras IA, Kremen SA, et al. False reports from patients with frontotemporal dementia: delusions or confabulations? Behav Neurol. 2011;24(3):237-244. PubMed CrossRef
29.Omar R, Sampson EL, Loy CT, et al. Delusions in frontotemporal lobar degeneration. J Neurol. 2009;256(4):600-607. PubMed CrossRef
30.Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266(8):2075-2086. PubMed CrossRef
31.Esterberg ML, Trotman HD, Holtzman C, et al. The impact of a family history of psychosis on age-at-onset and positive and negative symptoms of schizophrenia: a meta-analysis. Schizophr Res. 2010;120(1-3):121-130. PubMed CrossRef
32.Derntl B, Seidel E-M, Schneider F, et al. How specific are emotional deficits? a comparison of empathic abilities in schizophrenia, bipolar and depressed patients. Schizophr Res. 2012;142(1-3):58-64. PubMed CrossRef
33.Cooper JJ, Ovsiew F. The relationship between schizophrenia and frontotemporal dementia. J Geriatr Psychiatry Neurol. 2013;26(3):131-137. PubMed CrossRef
34.Bland AR, Roiser JP, Mehta MA, et al. EMOTICOM: a neuropsychological test battery to evaluate emotion, motivation, impulsivity, and social cognition. Front Behav Neurosci. 2016;10:25. PubMed CrossRef
35.Ducharme S, Pearl-Dowler L, Gossink F, et al. The Frontotemporal Dementia versus Primary Psychiatric Disorder (FTD versus PPD) Checklist: a bedside clinical tool to identify behavioral variant FTD in patients with late-onset behavioral changes. J Alzheimers Dis. 2019;67(1):113-124. PubMed CrossRef
36.Ghetti B, Oblak AL, Boeve BF, et al. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41(1):24-46. PubMed CrossRef
37.Wilhelmsen KC, Lynch T, Pavlou E, et al. Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21-22. Am J Hum Genet. 1994;55(6):1159-1165. PubMed
38.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5‘ ²-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702-705. PubMed CrossRef
39.Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815-825. PubMed CrossRef
40.Deleon J, Miller BL. Frontotemporal dementia. In: Geschwind DH, Paulson HL, Klein C, eds. Handbook of Clinical Neurology. Vol. 148. Amsterdam, Netherlands: Elsevier; 2018:409-430.
41.Woollacott IOC, Rohrer JD. The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem. 2016;138(suppl 1):6-31. PubMed CrossRef
42.Bardai FH, Wang L, Mutreja Y, et al. A conserved cytoskeletal signaling cascade mediates neurotoxicity of FTDP-17 tau mutations in vivo. J Neurosci. 2018;38(1):108-119. PubMed
43.Goedert M, Spillantini MG. Tau mutations in frontotemporal dementia FTDP-17 and their relevance for Alzheimer’s disease. Biochim Biophys Acta. 2000;1502(1):110-121. PubMed CrossRef
44.Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat. 2004;24(4):277-295. PubMed CrossRef
45.Baillon S, Murray J. A national survey of psychiatrists’ attitudes towards the physical examination [published online ahead of print January 11, 2019]. J Ment Health. PubMed
46.Azzam PN, Gopalan P, Brown JR, et al. Physical examination for the academic psychiatrist: primer and common clinical scenarios. Acad Psychiatry. 2016;40(2):321-327. PubMed CrossRef
47.Garden G. Physical examination in psychiatric practice. Adv Psychiatr Treat. 2005;11(2):142-149. CrossRef
48.Vijverberg EGB, Wattjes MP, Dols A, et al. Diagnostic accuracy of MRI and additional [18F]FDG-PET for behavioral variant frontotemporal dementia in patients with late onset behavioral changes. J Alzheimers Dis. 2016;53(4):1287-1297. PubMed CrossRef
This PDF is free for all visitors!



