ABSTRACT
Although antipsychotics have been available for almost 70 years and greatly improved outcomes for individuals with schizophrenia, all currently available options derive their efficacy from blockade of dopaminergic receptors. However, this mechanism of action leaves many symptoms unresolved and is associated with a significant side effect burden. The mechanisms underlying schizophrenia, which were initially thought to be related to excessive presynaptic dopamine in specific areas of the brain, are now understood to be much more complex and involve structural and molecular changes throughout brain circuits. Consequently, drug discovery efforts have sought new targets in the search for safer and more effective medications that can improve symptoms of schizophrenia and psychosis, including trace amine–associated receptors (TAARs), muscarinic receptors, and serotonergic receptors. Positive phase 2 trial results indicating efficacy and safety of the TAAR1 agonist ulotaront (SEP-363856) and of the muscarinic M1/M4 agonist KarXT (xanomeline plus trospium) for total, positive, and negative symptoms in patients with acute exacerbation of schizophrenia, and of the serotonin 5-HT2A agonist/antagonist pimavanserin in patients with schizophrenia and predominant negative symptoms for negative symptom control are encouraging. Taken together, these data indicate in the context of ongoing phase 3 trial programs that patients with schizophrenia may soon have access to the first non–D2 blocking medication, which could drastically change the treatment landscape and improve outcomes for many of the individuals with schizophrenia who do not fully respond to or cannot tolerate currently available antipsychotic agents that currently all act via postsynaptic dopamine D2 receptor blockade.
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
J Clin Psychiatry 2022;1(InfoPack 1):SU21024IP1
To cite: Correll CU, Abi-Dargham A, Howes O. Emerging treatments in schizophrenia. J Clin Psychiatry 2022;1(InfoPack 1):SU21024IP1.
To share: https://doi.org/10.4088/JCP.SU21024IP1
© Copyright 2022 Physicians Postgraduate Press, Inc.
Faculty
 |
 |
 |
|---|---|---|
| Christoph U. Correll, MD, Chair | Anissa Abi-Dargham, MD | Oliver Howes, MRCPsych, PhD |
| Department of Psychiatry, The Zucker Hillside Hospital, Glen Oaks, New York; Department of Psychiatry and Molecular Medicine, Donald and Barbara Zucker School of Medicine at Hofstra/Northwell, Hempstead, New York; and Department of Child and Adolescent Psychiatry, Charité Universitätsmedizin, Berlin, Germany | Department of Psychiatry, Renaissance School of Medicine, Stony Brook University, New York | Institute of Psychiatry, Psychology and Neuroscience, King’s College, London, UK |
Acknowledgment
This InfoPack is based on a series of teleconferences by 3 experts on schizophrenia. These teleconferences were held in October 2021. This evidence-based, peer-reviewed InfoPack was prepared by Healthcare Global Village, Inc. Financial support for the preparation and dissemination of this InfoPack was provided by Sunovion Pharmaceuticals Inc. The opinions expressed herein are those of the authors and do not necessarily reflect the views of Healthcare Global Village, Inc; the publisher; the American Society of Clinical Psychopharmacology; or Sunovion Pharmaceuticals Inc.
In the April 16, 2020, issue of The New England Journal of Medicine,1 Koblan and colleagues published their findings from a phase 2 clinical trial of ulotaront (SEP-363856) in adults experiencing an acute exacerbation of schizophrenia. This trial found ulotaront to be significantly more effective than placebo for reducing Positive and Negative Syndrome Scale (PANSS) scores. This study has been met with great interest because, unlike all other available antipsychotic agents, ulotaront does not act on dopamine D2 receptors. This InfoPack will begin with a summary of the trial conducted by Koblan et al, followed by an analysis by Drs Anissa Abi-Dargham, Oliver Howes, and Christoph U. Correll in which they put these data into the context of available drugs to treat psychosis, brain circuits thought to be involved in psychosis, and agents in phase 3 trial programs that aim to ameliorate psychotic symptoms without directly blocking postsynaptic dopamine receptors.
Summary
A Promising New Treatment Target: Overview of a Randomized Controlled Trial of Ulotaront (SEP-363856) for Acute Exacerbation of Schizophrenia
Full article online from N Engl J Med 2020;382(16):1497–1506
Although several new antipsychotics have become available in the 7 decades since the introduction of chlorpromazine, all of these treatments have relied primarily on the same mechanism—blockade of postsynaptic dopamine D2 receptors. Because of the complex and heterogeneous nature of schizophrenia, discovery of new therapeutic targets has proven to be very difficult. Using an in vivo phenotypic approach, investigators were able to identify a new compound that exhibited behavioral effects consistent with an antipsychotic effect. This compound, SEP-363856 (since named ulotaront by the WHO classification system), has agonist activity at trace amine–associated receptor 1 (TAAR1) and 5-hydroxytryptamine type 1A (5-HT1A [serotonin]) receptors but with absence of D2 and 5-HT2A receptor blockade.2 Following is an overview of the phase 2 trial of ulotaront,1 which was originally published in The New England Journal of Medicine.
Trial methods. The trial was conducted from December 2016 through July 2018 across 34 sites in Eastern Europe and North America. The trial design is summarized in Figure 1. The active treatment phase was preceded by a screening and washout period of up to 14 days, during which all psychotropic medications were discontinued. Patients were then randomly assigned in a 1:1 ratio to receive either placebo or a flexible dose of ulotaront once daily at bedtime. No one involved in any aspect of the trial was aware of the treatment assignments. The initial dose of ulotaront was 50 mg with an option to increase to 75 mg on or after day 4. The dose could be reduced to 50 mg at any point if tolerability issues arose. The trial lasted for 4 weeks.
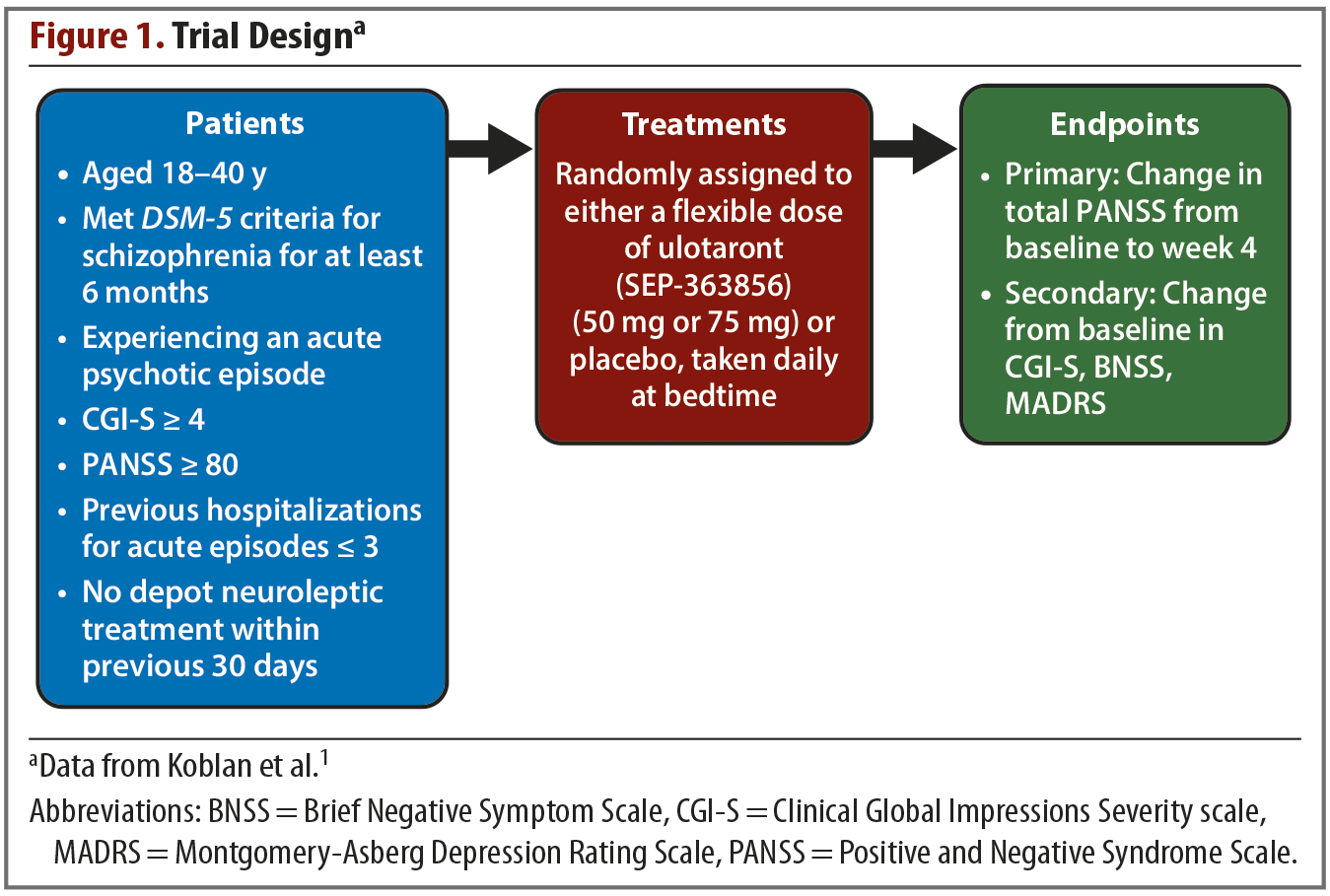
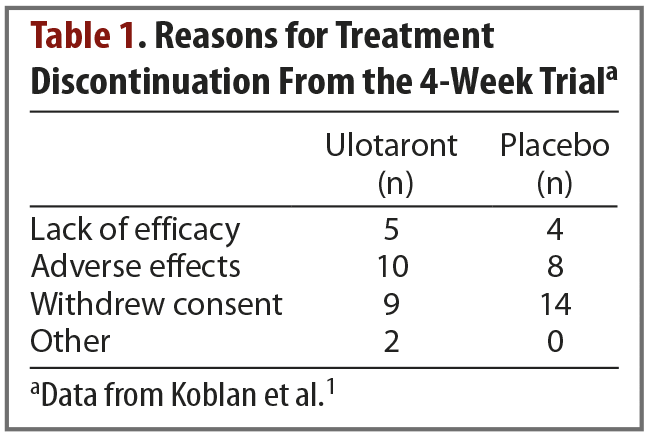
Results. The trial began with 245 enrolled patients—120 in the active treatment group and 125 in the placebo group. A total of 52 patients discontinued treatment, and they were split evenly between the active treatment and placebo arms. Reasons for treatment discontinuation are listed in Table 1. The primary efficacy measure was change from baseline in total PANSS score. Compared with patients receiving placebo, those receiving ulotaront experienced significantly greater reduction in total PANSS scores (–17.2 vs –9.7; P = .001). Patients in the ulotaront group also showed greater improvements in all secondary efficacy measures. The incidence of side effects was comparable between the ulotaront and placebo groups, with the most common being somnolence, agitation, and nausea. Patients taking ulotaront gained an average of 0.3 kg in body weight, while those taking placebo lost an average of 0.1 kg. Total and low-density lipoprotein (LDL)–cholesterol levels decreased in patients taking ulotaront (no change in the placebo group). No other changes in metabolic measures were observed. The incidence of extrapyramidal symptoms was comparable in both groups.
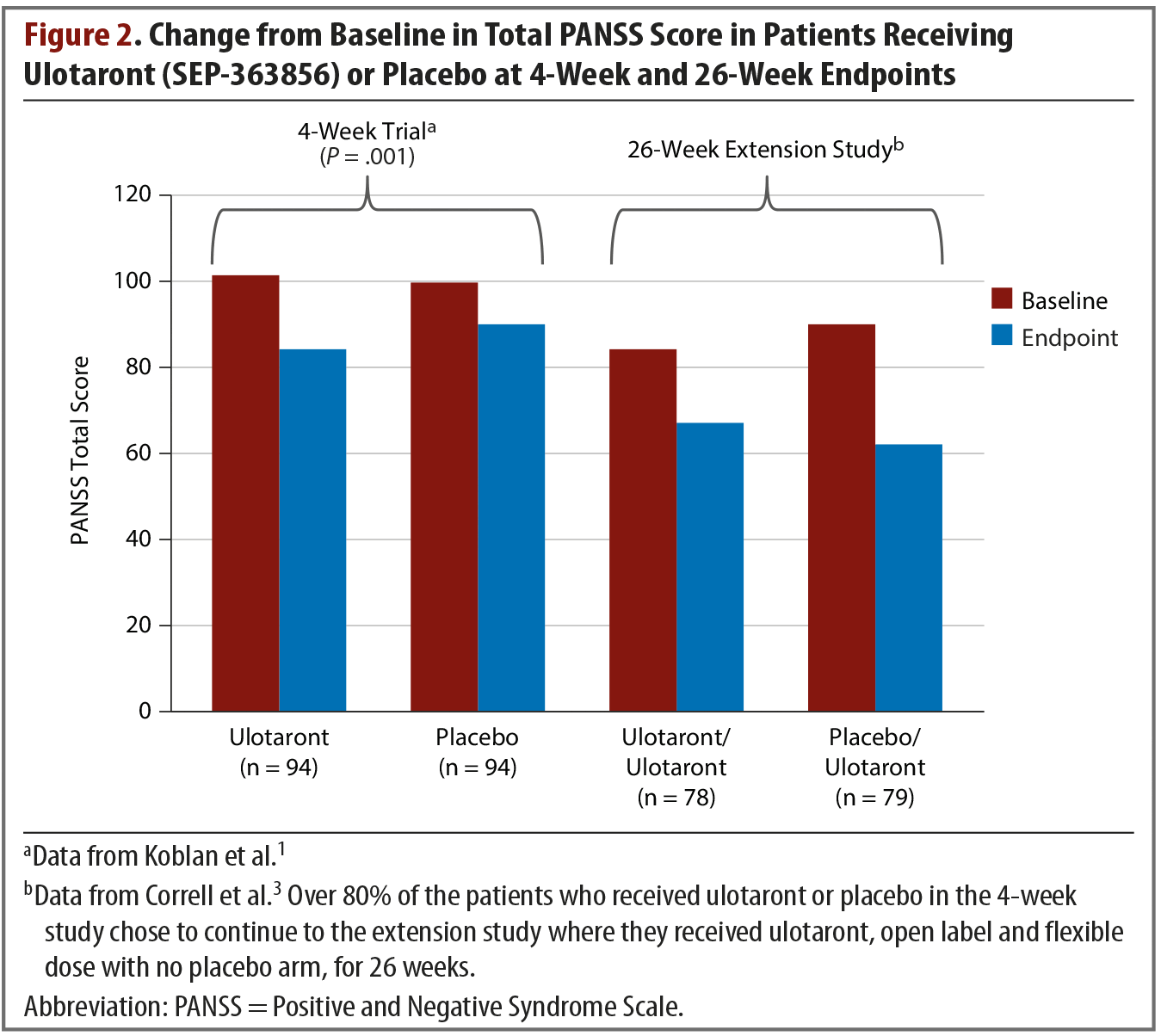
Extension study. At the conclusion of the 4-week trial, 157 patients opted to enter a 26-week, open-label, extension study.3 They received flexible doses of ulotaront of 25 mg, 50 mg, or 75 mg per day. At endpoint, the total PANSS score of patients who had received ulotaront in the initial trial and who were then treated open label for another 26 weeks had been reduced by an additional average of 17.1 points in addition to the acute trial improvements (Figure 21,3). The patients who had received placebo during the 4-week trial and then switched to ulotaront for the extension study exhibited a mean reduction in total PANSS score of 27.9. This combined group’s cumulative change in total PANSS score from study entry to extension study endpoint was –37.6. Approximately 9% of patients experienced serious adverse events related to mental health, including schizophrenia exacerbations, psychosis, depression, and suicidal ideation. At the 26-week endpoint, with all patients taking ulotaront, the pooled weight change was –0.3 kg, and lipid measures showed decreases or no change.3 Additional adverse effects reported were generally mild, with headache and insomnia being most common.
Conclusions
In this phase 2 acute, double-blind, placebo-controlled trial and open-label extension study, treatment with ulotaront was associated with significant improvements in total PANSS score compared with placebo and was generally well tolerated. Ulotaront appears to be a promising new treatment, but larger trials are needed.
Commentary
The Pathophysiology of Schizophrenia: A Brief Overview
Anissa Abi-Dargham, MD
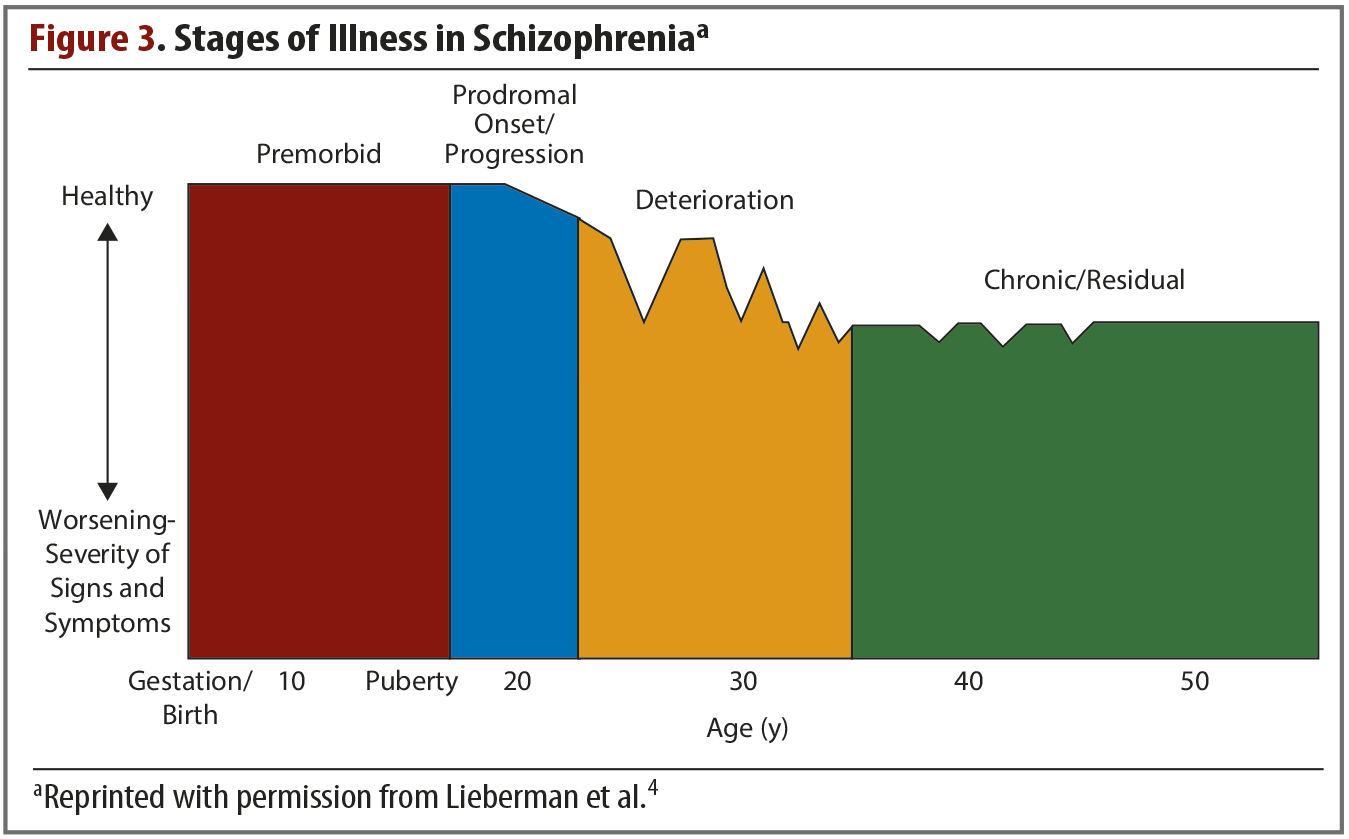
Schizophrenia is a chronic disorder in which symptoms typically emerge in late adolescence or early adulthood. For most patients, these symptoms will progress through several clinical stages of worsening severity (Figure 3),4 and they occur in clusters across multiple functional domains. These symptom clusters, which have been observed since schizophrenia was first identified as a distinct disorder, include deficits and dysfunction in perception, cognition, mood, movement, behavior, and reward.4 Although clinical symptoms typically do not emerge until adolescence or early adulthood, decades of investigation have shown that they stem from widespread brain alterations that begin very early in life. Schizophrenia is thought of as a global brain disease, and a greater knowledge of the abnormalities that occur in different systems will likely lead to more effective, targeted treatments.
Neurodevelopmental Changes
Evidence indicates that environmental and genetic factors may affect brain development in utero, conferring vulnerability to the development of schizophrenia. These factors alter the brain’s developmental trajectory leading to abnormalities in different circuits and transmitter systems.4 Typically, synapse proliferation and migration occur from gestation through the first year of life, followed by a period of synaptic refinement and myelination lasting until early adulthood.5 Excitatory synapses are pruned, while inhibitory synapses proliferate and develop connections, resulting in an intricate balance of excitation and inhibition. In schizophrenia, excessive pruning of excitatory synapses and a deficient development of inhibitory synapses are thought to result in an imbalance of excitation and inhibition. This imbalance has been a major focus of research in schizophrenia.
Imaging studies have provided evidence that schizophrenia is associated with structural changes in both gray and white matter. A meta-analysis of 27 magnetic resonance imaging (MRI) studies detected widespread gray matter decreases in the hippocampus, the insula, the anterior cingulate, various regions of the frontal cortex, the basal ganglia, and the thalamus.6 These gray matter decreases may be the result of alterations at the cellular level. Postmortem studies have revealed abnormalities in circuitry in the prefrontal cortex (PFC) that may account for decreases in brain volume.7 Patients with schizophrenia have been found to have decreases in somal size and dendritic spine density of pyramidal cells in the PFC. Furthermore, reductions in the enzyme that synthesizes GABA (γ-aminobutyric acid), as well as decreases in the GABA transporter levels, have been observed; these may contribute to decreases in gray matter volume and may lead to the imbalance of GABAergic and glutamatergic transmission that is needed for optimal cognitive function. White matter changes in schizophrenia have been investigated using diffusion tensor imaging, revealing abnormalities in frontotemporal white matter connectivity between certain cortical regions including the frontal, temporal, and genu of the corpus callosum regions.8 White matter tract abnormalities are associated with severity of clinical symptoms.
MRI has revealed abnormalities associated with schizophrenia in additional parts of the brain. An imaging study9 by Schobel and colleagues examined cerebral blood volume (CBV) in patients with schizophrenia, patients at high risk for developing schizophrenia, and healthy controls. Enhanced CBV, which indicates areas of neuronal overactivity, was found in the CA1 subfield of the hippocampus in both the schizophrenia and high-risk groups. CBV, therefore, could be useful as a biomarker to identify patients who will progress to schizophrenia, although a replication of this finding has not yet been reported. The auditory cortex is also overactive in schizophrenia. A functional (f)MRI study10 found that the auditory cortex in patients with schizophrenia is hyperactive during silence, and this hyperactivity is associated with the severity of auditory hallucinations.
Neurochemical Changes
Advances in imaging technology have allowed investigation of the neural circuitry underlying schizophrenia. Normal brain function requires information processing through interactions among different neural networks.11 In particular, the basal ganglia-thalamocortical (BGTC) network controls the flow of information from the cerebral cortex, into striatum, pallidum, and thalamus, feeding back into the cortex.12 These BGTC loops receive inputs from dopaminergic projections arising from the midbrain, including ventral tegmental area and substantia nigra; they have a major influence in organizing behavior across different functional domains. (See Figure 6.)
Within the BGTC network, output from the striatum consists in medium spiny GABAergic projection neurons. These neurons are distinguished in 2 pathways according to whether they have D1 or D2 receptors. The D1 pathway is considered the “go” pathway, or direct pathway, and it facilitates communication between the thalamus and the cortex. The D2 pathway is the “no-go” pathway, or indirect pathway, and it inhibits thalamocortical communication. The balance between “go” and “no-go” pathways is involved in determining approach and avoidance behavior across the different domains of function13 subserved by these BGTC loops. This important functional network is thought to be disrupted in schizophrenia, which leads to cognitive and behavioral symptoms. Although a number of neurotransmitters are involved in the processing of information through neural networks, one main dysfunction is thought to stem from dopaminergic abnormalities.12
Dopamine. Dopamine is critical for understanding schizophrenia and has been a major focus of research into the pathophysiology of the illness and treatment development. The earliest evidence of dopamine’s involvement in schizophrenia was based on clinical observations, such as the observation that antipsychotic drugs that bind to D2 receptors were effective at reducing positive symptoms.12 Similarly, dopamine agonists were found to induce or worsen psychosis. These observations led to the hypothesis, which has evolved over time, that psychotic symptoms stem from an excess of dopamine in the striatum (Figure 4).14
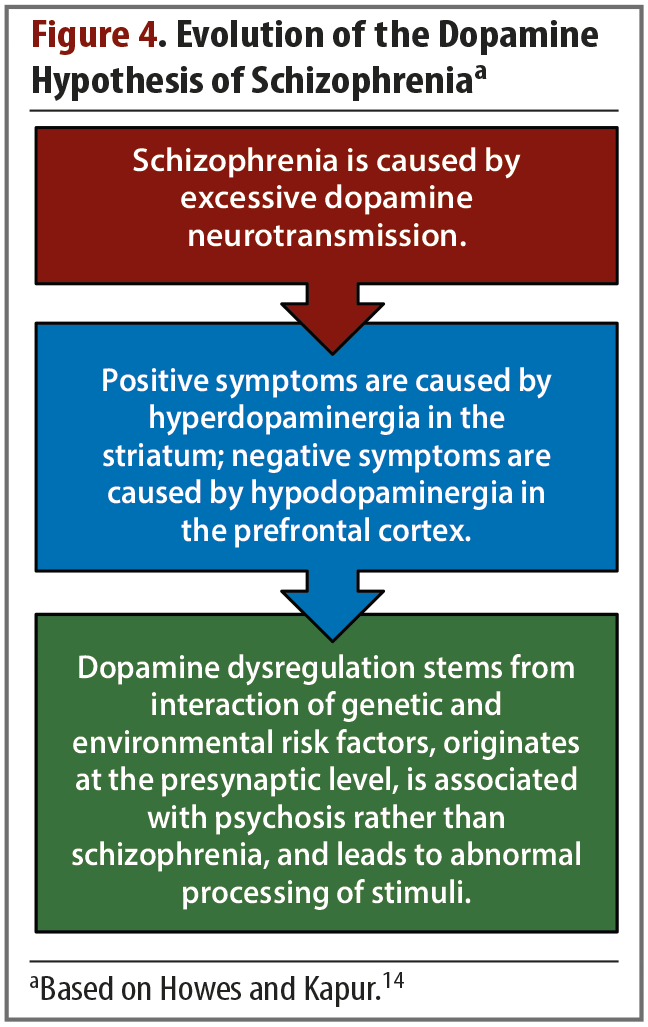
Once imaging technology was available to investigate dopaminergic activity in the striatum, studies were able to detect a number of schizophrenia-related changes to the striatal dopamine synaptic machinery, namely elevations in presynaptic dopamine synthesis capacity, dopamine release, and dopamine D2 receptor density.14 More specifically, dopamine hyperactivity is most pronounced in the associative regions of the striatum and is evident beginning in the prodromal stage of schizophrenia.15,16 This increase in dopamine in the associative striatum is thought to be the basis of positive symptoms. In the ventral striatum, low levels of dopamine relate to negative symptoms, while in the associative striatum, high levels of dopamine relate to psychotic symptoms.17
The manner in which striatal dopaminergic dysfunction manifests as positive and negative symptoms is related to dopamine’s role in learning through salience processing and reward prediction.17 Research shows that the firing pattern of midbrain dopamine neurons tracks the mismatch between expectations of rewards and the experience of rewards. If a reward is the same as what was expected, dopamine firing remains stable, but if the reward is better than expected, dopamine firing increases, and if the reward is worse than expected, dopamine firing is diminished. Both of these processes are important to reinforce learning and guide future behavior.18
In patients with schizophrenia, dopamine cells may fire unpredictably in response to stimuli and rewards.19 If dopamine cells fire in response to insignificant stimuli, thus investing those with unwarranted meaning, this may form the basis for delusional thinking. Conversely, individuals with schizophrenia show a blunted dopaminergic response to reward anticipation, which may correspond to negative symptoms.17
Investigation into dopamine dysfunction in schizophrenia progressed from the original hypothesis that focused on striatal dopamine to a more complex framework, which acknowledged that dopamine dysfunction was spread throughout different regions of the brain.7 Five dopaminergic pathways (includes 2 nigrostriatal pathways) have been identified that are involved in the symptoms and pathology of schizophrenia (Figure 5).20 All 5 of these pathways begin in the midbrain, in either the ventral tegmental area or the substantia nigra. From the midbrain, dopamine projects to regions of the cortex, thalamus, and striatum. This functional connectivity is integral to the BGTC functional network. In schizophrenia, abnormal dopamine transmission in the striatum may relate to abnormal functional connectivity of the striatal substructures to the rest of the brain.12
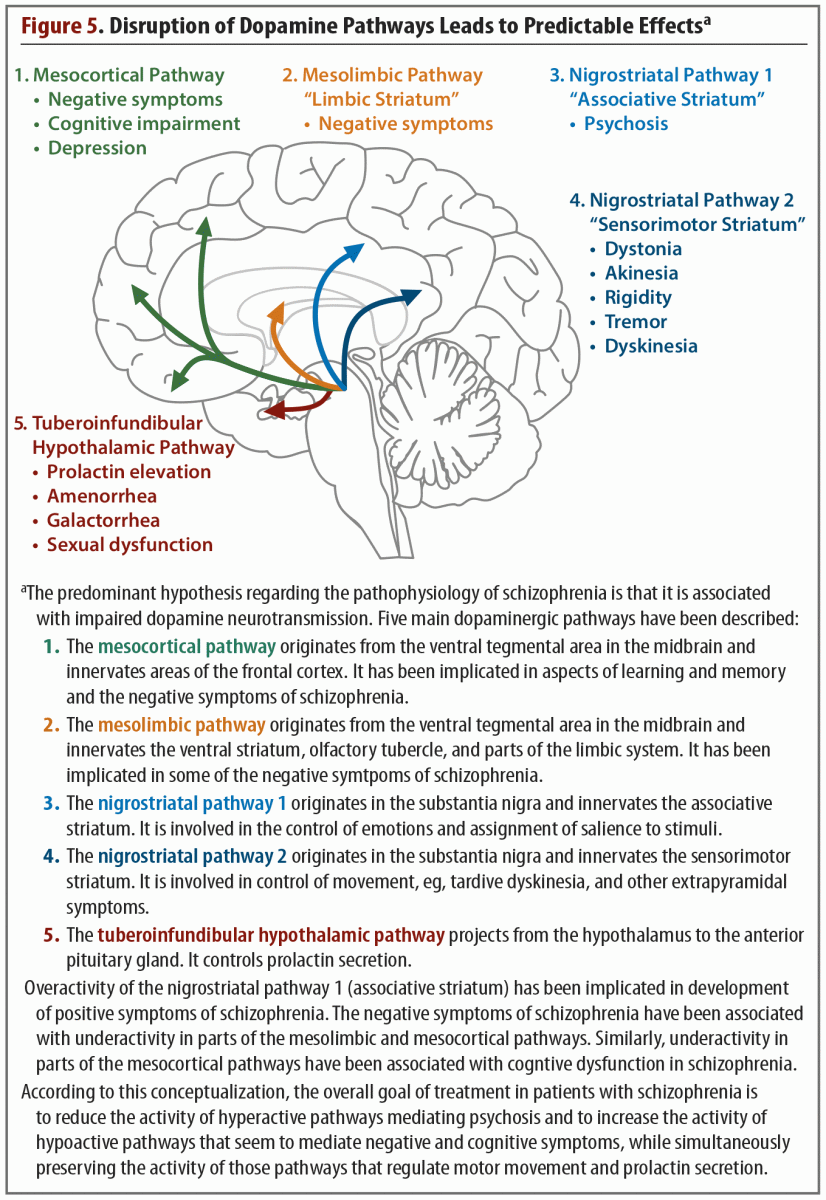
Dopaminergic dysfunction in cortical regions is now understood to be a central part of the pathophysiology of schizophrenia. Dopaminergic receptors in the PFC maintain a balance of excitation/inhibition by modulating glutamate neurotransmission and GABAergic interneuron function.21 This microcircuitry matures during adolescence, achieving optimal stimulation of the pyramidal cells and the GABAergic interneurons by dopamine receptors and resulting in more efficient and fine-tuned cortical activity. In schizophrenia, this maturation does not occur, leading to functional and behavioral deficits.21
Cortical dopamine release is thought to be deficient in patients with schizophrenia. Slifstein and colleagues22 conducted an imaging study to observe amphetamine-induced dopamine release in the dorsolateral PFC of 20 patients with schizophrenia and 21 healthy controls. Compared with controls, individuals with schizophrenia showed reduced dopamine release not only in the dorsolateral prefrontal cortex (DLPFC), but also in most cortical and extrastriatal regions. Furthermore, the diminished dopamine release capacity in the DLPFC of patients was correlated with poor performance on a working memory task, as reported by Cassidy and colleagues.23
In summary, a great deal of evidence supports the involvement of dopaminergic dysfunction in the pathophysiology of schizophrenia. Excess dopamine has been observed in the striatum, but deficit of dopamine has been observed everywhere else, in particular, the regions important for executive function, working memory, and social cognition.
Glutamate. Dysfunction of the glutamatergic system is also thought to be involved in the pathophysiology of schizophrenia. Glutamate is the main excitatory neurotransmitter in the central nervous system, and glutamatergic neurons are found throughout the brain. Numerous types of glutamate receptors exist, classified as either ionotropic or metabotropic. Ionotropic receptors can be further classified as either N‐methyl‐d‐aspartate (NMDA) receptors or non-NMDA receptors. Ionotropic receptors affect the firing pattern of dopaminergic neurons in the midbrain.24 Glutamatergic dysfunction was first hypothesized to contribute to schizophrenia after it was discovered that the administration of NMDA antagonists led rodents and non-human primates to exhibit behavior reminiscent of schizophrenia-like symptoms in humans.25 In humans, healthy controls who have been administered phencyclidine or ketamine, both of which are NMDA receptor antagonists, have been found to exhibit behaviors similar to the positive, negative, and cognitive symptoms of schizophrenia.
NMDA receptor antagonists such as ketamine inhibit NMDA receptors on GABA interneurons, leading to reductions in extracellular GABA levels and increases in extracellular glutamate levels, resulting in increased cortical activity. An fMRI study26 was conducted in which healthy volunteers were given an infusion of ketamine in order to investigate changes in functional brain connectivity and the manner in which these changes might relate to symptoms. Global functional hyperconnectivity was observed after 45 minutes of continuous administration. Region-specific changes in global brain connectivity were identified that predicted positive and negative symptoms, and these correspond to the abnormalities in functional connectivity that have been observed in schizophrenia.
Studies have used proton magnetic resonance spectroscopy (1H-MRS) to examine glutamate in different brain regions in schizophrenia, particularly the frontal cortex.27 A meta-analysis was conducted of 1H-MRS imaging studies that compared glutamate and glutamine concentrations in the brains of people with schizophrenia and healthy controls. Glutamine is a metabolite of glutamate and is considered an indicator of glutamate neurotransmission.25 Compared with healthy controls, patients with schizophrenia were found to have higher concentrations of glutamine and lower concentrations of glutamate in the frontal region. These levels of both glutamate and glutamine were found to decrease with age. Thus, abnormalities in the glutamatergic system change as the illness progresses. Early in the illness, increased glutamate in the frontal cortex is associated with severity of positive symptoms, but with time and exposure to medications, glutamate decreases, as do positive symptoms.
Schizophrenia is a multifactorial disease, involving genetic and environmental factors, as well as abnormalities in brain development, circuitry, and synchrony. These abnormalities occur on structural, functional, and molecular levels and have widespread effects on cognitive functioning.
—Dr Abi-Dargham
Additional neurotransmitter dysfunctions. The pathophysiology of schizophrenia is increasingly understood as stemming from dysfunction in multiple interconnected neurotransmission circuits. Abnormalities in the cholinergic system that have been observed in schizophrenia include a reduced number of cholinergic interneurons in the striatum, reduced numbers of M1 and M4 receptors in the caudate and putamen, and reduced muscarinic receptor availability in the cortex and basal ganglia.28 Cholinergic projections from the brainstem extend to the midbrain where they stimulate dopamine neurons in the substantia nigra and ventral tegmental area. This leads to dopamine release along the mesolimbic pathway and increased dopamine in the striatum.28 The cholinergic system also interacts with the glutamatergic and GABAergic systems, thus suggesting that any dysfunction in acetylcholine neurotransmission could disrupt the balance of excitation and inhibition maintained by glutamate and GABA signaling.29
Trace amine–associated receptors were only discovered in 2001, and although much has been discovered regarding their structure, location, and function, a great deal remains unknown.30 TAARs are distributed throughout the body, and they can be activated by not only trace amines, but also monoamines such as dopamine or serotonin, meaning they have the potential to modulate these neurotransmitters. A number of TAARs have been identified, but TAAR1 has been studied most extensively and is the most widespread TAAR in the brain. They are G-protein–coupled receptors and are mostly located intracellularly, but they can transfer to the plasma membrane after creating a heterodimer with another receptor.31 Of particular relevance to schizophrenia, TAAR1 has the ability to undergo heterodimerization with plasma membrane D2 receptors leading to internalization of both receptors, thereby reducing dopamine expression,32 and is also positioned to modulate serotonergic and glutamatergic signaling33 Genetic variations in TAAR1 have been observed in individuals with schizophrenia that may contribute to dopaminergic dysfunction.34
Conclusions
Schizophrenia is a complex disorder that is still not fully understood. Decades of research have revealed neurodevelopmental, structural, functional, and molecular abnormalities that all contribute to positive, negative, and cognitive symptoms and that may provide targets for future treatments.
Current Paradigm for Schizophrenia Treatment: D2 Receptor Blockade
Oliver Howes, MRCPsych, PhD
The first antipsychotic drug was discovered almost 70 years ago.35 In the intervening years, more than 20 different antipsychotics have been developed, and this high number would seem to indicate that patients and providers have many treatment options from which to choose. However, all of these agents are postsynaptic D2 blockers, meaning that treatment options are actually quite limited. With the possible exception of clozapine, all available antipsychotics have the same core mode of action,36 and, despite variations in non-dopaminergic transmitter interactions, they all have similar benefits and also many of the same drawbacks associated with D2 blockade.
Evidence of Antipsychotic D2 Receptor Blockade
In the 1970s, 2 landmark in vitro studies37,38 were able to demonstrate the link between D2 receptor affinity and antipsychotic clinical potency in vitro. Positron emission tomography (PET) imaging has allowed researchers to observe the effects of antipsychotics on D2 receptors in vivo. In a study of 22 first-episode patients with schizophrenia being treated with haloperidol, Kapur and colleagues39 used PET imaging and observed that higher D2 receptor occupancy within the first week of treatment predicted greater subsequent clinical response. The finding that symptom reduction can be achieved through blockade of D2 receptors fits with existing knowledge of the neurobiology of schizophrenia. Abnormalities in presynaptic aspects of the dopamine system, in particular the synthesis and release capacity of dopamine, occur in the striatum, which leads to delusions and other positive symptoms. Therefore, blocking the D2 receptors will alleviate these symptoms, but as the D2 receptors are predominantly postsynaptic, these actions are downstream from the dopamine dysfunction seen in the disorder, which is predominantly presynaptic.25
This dopamine receptor blocking–based paradigm is the basis of all existing antipsychotic treatment. Numerous placebo-controlled trials conducted over the past 60 years have shown that all available antipsychotics can reduce positive symptoms. Leucht and colleagues40 conducted a meta-analysis of 167 double-blind, randomized controlled trials of 15 different first- and second-generation antipsychotics and found that twice as many patients improved with antipsychotic treatment compared with those taking placebo.
Limitations of Existing Treatments
Treatment discontinuation. One of the greatest drawbacks of current antipsychotics is that they are associated with high rates of discontinuation. The European First Episode Schizophrenia Trial (EUFEST)41 found discontinuation rates of between one-third to two-thirds of patients during the first year of treatment. Haloperidol was associated with the highest discontinuation rate (72%) and olanzapine with the lowest (33%). Furthermore, treatment discontinuation is also a problem for patients with chronic schizophrenia. A study42 conducted as part of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) randomly assigned 444 patients who had already discontinued treatment with one second-generation antipsychotic to begin treatment with a different second-generation antipsychotic. This study found that 77% of patients had discontinued treatment with the new antipsychotic over the course of 12 months, highlighting high rates of treatment discontinuation as a major clinical challenge.
Inefficacy. Lack of efficacy is a common reason for treatment discontinuation.41,42 Although available antipsychotics can alleviate positive symptoms through blocking striatal dopamine D2 receptors, a considerable number of patients fail to show improvement in psychotic symptoms with these agents.36 Studies have found rates of treatment resistance to range from 23% in first-episode patients43,44 to 56% in a community sample of chronic patients.45 The hypothesis was put forth that these patients were not responding to treatment because sufficient D2 receptor blockade was not being achieved, but PET studies have shown comparable dopamine D2 receptor occupancy in both treatment-responsive and treatment-resistant patients, indicating that inadequate D2 receptor blockade does not explain treatment resistance in these patients.46
Current antipsychotic treatments are also largely ineffective for the negative and cognitive symptoms of schizophrenia.47 These symptoms are thought to stem from impaired neural communication caused by disruptions in brain networks, leading to an imbalance of excitatory and inhibitory cortical activity and low dopamine neurotransmission in the cortex and limbic regions. Thus, postsynaptic dopamine D2 receptor blockade would not affect these processes. Moreover, some evidence indicates that the D2 blockade might actually be associated with worsening of negative and cognitive symptoms.36 For example, healthy volunteers have been found to exhibit negative symptoms and impairments in cognitive functioning after being administered antipsychotic medications.48,49
Because more than 20 different antipsychotics are currently available, clinicians appear to have many options to offer patients, but because they are all D2 receptor blockers, clinicians really have very little to offer to patients who do not improve or cannot tolerate their current antipsychotic.
—Dr Howes
Tolerability. Dopamine D2 receptor occupancy is also related to many of the side effects that often lead to treatment discontinuation. Extrapyramidal side effects, such as akathisia, parkinsonism, and tardive dyskinesia, become more common as dopamine D2 receptor occupancy exceeds about 80%, and hyperprolactinemia increases at D2 receptor occupancy above about 75%.36 The raised prolactin levels associated with antipsychotic treatment can then lead to sexual side effects.50
Available antipsychotics are associated with other troubling side effects that are thought to stem from actions at other receptors.36 Sedation is most common with clozapine, quetiapine, olanzapine, and chlorpromazine, and these antipsychotics have high affinities for histamine H1 receptors. Anticholinergic effects, such as dry mouth and gastrointestinal disturbance, have been associated with clozapine and olanzapine, which have high affinity for postsynaptic muscarinic receptors.
Perhaps the most problematic side effects of antipsychotic treatment are weight gain and metabolic disturbances. These issues require special attention because they can affect a patient’s long-term health. A meta-analysis51 of over 100 randomized controlled trials, which included more than 25,000 patients, found that even short-term treatment of acute episodes could lead to significant increases in body weight, LDL cholesterol, and glucose levels, any of which can increase a patient’s risk of cardiovascular disease in the long-term. These side effects may be in part the indirect result of dopamine D2 receptor blockade as well as the off-target actions at other receptors.36 Dopamine receptors are involved in reward signaling, and, therefore, blocking D2 receptors may disrupt these reward signals as well as signals of satiety following food consumption and lead to overeating. Overall, one can conclude that the D2 receptor blocking paradigm has many limitations both in terms of efficacy and side effects, highlighting the need for alternative approaches.
New Mechanisms of Action for the Treatment of Schizophrenia
Christoph U. Correll, MD
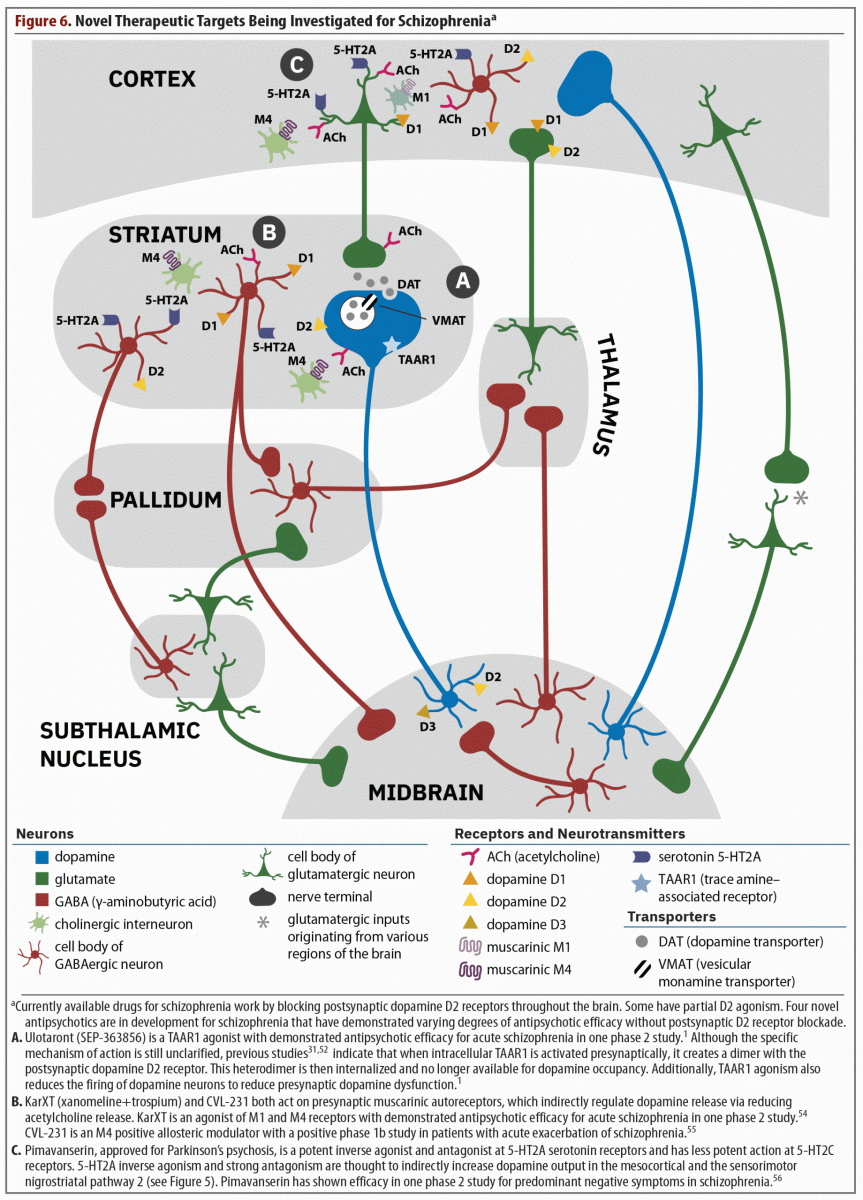
That only postsynaptic dopamine blockers are available to treat schizophrenia is quite detrimental for this population because of the adverse effects and inefficacy commonly associated with these agents. Improved treatments are needed that are able to control residual positive, negative, and cognitive symptoms while not worsening affective, motor, and cardiometabolic domains. Currently, 4 medications have shown promise for the treatment of symptoms in schizophrenia without blocking the postsynaptic dopamine receptor (Figure 6).1,31,52,54–56
Ulotaront
As discussed previously in the overview of the article by Koblan et al,1 ulotaront is a TAAR1 and 5-HT1A agonist, and although the specific mechanism of action has not been fully elucidated, agonism at these 2 receptors contributes to its efficacy.2 Two review articles31,52 note that when the TAAR1 receptor is stimulated, several important events occur. These include a dimerization of the TAAR1 receptor with the postsynaptic D2 receptor, which leads to internalization of the D2 receptor (see part A of Figure 6). Therefore, less D2 receptor occupancy is possible without actually blocking the D2 receptor. Furthermore, presynaptic firing of dopamine is also reduced and has been shown to reduce excess striatal dopamine synthesis in a model of the pathophysiology of schizophrenia.53 Thus, ulotaront might represent the first presynaptic treatment for schizophrenia. This development is noteworthy because this agent would be targeting one of the actual mechanisms underlying the pathophysiology of schizophrenia—the overproduction of dopamine—rather than a dysfunction of postsynaptic dopamine receptors that has not been clearly proven. Additionally, TAAR1 receptors also exist in the periphery and regulate appetite, reduce fasting glucose in the liver, increase insulin production, and delay gastric emptying, all of which contribute to improved glucose levels and metabolism.31
In the previously discussed phase 2 study published in the New England Journal of Medicine,1 ulotaront treatment for 4 weeks at doses of 50 or 75 mg/d was associated with significantly greater reductions in PANSS total scores, CGI-S scores, and all PANSS subscale scores compared with placebo. Also, although patients were not selected for having particularly high negative or depressive symptoms, those receiving ulotaront treatment also showed significantly more improvement on Brief Negative Symptom Scale and Montgomery-Asberg Depression Rating Scale scores compared with those receiving placebo. Few adverse effects emerged, with the most common being somnolence, agitation, and nausea, which ranged from 5%–7% with ulotaront and 3%–5% for placebo. Notably, weight gain was minimal at 0.3 kg in patients receiving ulotaront compared with –0.1 kg in patients receiving placebo. No appreciable effects on metabolic parameters were detected.
In the open-label extension study,3 157 (81.3%) of the 193 4-week completers continued into the open-label extension study, of whom 66.9% completed the 26-week study. In this 26-week study, ulotaront flexibly dosed at 25–75 mg continued to be associated with minimal changes in body weight (–0.3 ± 3.7 kg), lipid, glucose, and prolactin parameters. Movement disorder scales indicated no extrapyramidal effects. Moreover, continued improvements in change from open-label baseline in the PANSS total score of –22.6 were observed.
Xanomeline
Xanomeline is a muscarinic cholinergic receptor agonist that preferentially stimulates M1 and M4 receptors (see part B of Figure 6).54 The M4 receptor is mostly distributed in the brain, and stimulation of the M4 autoreceptor downstream reduces acetylcholine release at the interneuronal level, reducing dopamine transmission. Therefore, there is decreased dopamine transmission and firing without blockade of the postsynaptic dopamine receptor. In early trials of xanomeline for the treatment of Alzheimer’s disease and schizophrenia, patients experienced cholinergic side effects serious enough to halt investigation despite promising improvements in psychotic symptoms. When clinical development resumed, xanomeline was combined with trospium, a non–centrally active anticholinergic medication. This combination led to a clinically relevant reduction in the incidence of anticholinergic side effects during treatment with xanomeline.
In a phase 2B study by Brannan and colleagues,54 182 patients with schizophrenia were randomly assigned in a 1:1 ratio to receive either xanomeline-trospium or placebo for 5 weeks. Xanomeline was started at 50 mg twice a day combined with 20 mg of trospium and increased to 125 mg of xanomeline and 30 mg of trospium twice a day. Dosage could be reduced to 100 mg of xanomeline and 20 mg of trospium if tolerability issues arose. After 5 weeks, patients receiving xanomeline-trospium showed a significantly greater reduction in scores on both the PANSS total and subscale scores (P < .001). Patients in the active treatment group experienced more side effects than those in the placebo group, with the most common being constipation (17% vs 3% with placebo), nausea (17% vs 12% with placebo), dry mouth (9% vs 1% with placebo), dyspepsia (9% vs 4% with placebo), and vomiting (9% vs 4% with placebo). Despite these adverse events, very few people discontinued treatment. In terms of weight gain, patients in the xanomeline-trospium group gained an average of 1.5 kg compared with an increase of 1.1 kg in the placebo group. No appreciable changes in extrapyramidal side effects, akathisia, and metabolic parameters were observed.
Seventy years after the discovery of the first treatment for psychosis, 4 new treatments, each with a different mechanism of action, are being investigated for schizophrenia and show promise for greater efficacy and tolerability than currently available antipsychotics.
—Dr Correll
CVL-231
CVL-231 is an M4 positive allosteric modulator that is thought to prevent acetylcholine release by activating M4 autoreceptors (see part B of Figure 6).55 Without acetylcholine, dopamine output decreases, potentially reducing psychotic symptoms. CVL-231 was investigated in a phase 1B trial that was mainly designed to be a dose-finding and pharmacokinetic study. The 81 enrolled patients were randomized at 27 patients each to either placebo, 30 mg of CVL-231 once per day, or 20 mg of CVL-231 twice a day. Although this study is limited by its small sample size, at the end of the 6-week study, both doses of CVL-231 were associated with significantly greater reductions vs placebo in total PANSS score (CVL-231 30 mg qd: P = .023; CVL-231 20 mg bid: P = .047) and PANSS negative symptom scale scores (CVL-231 30 mg qd: P = .009; CVL-231 20 mg bid: P = .002).57 In terms of positive symptoms, only the 30-mg, once-daily dose was significantly superior to placebo (P = .016). Few side effects emerged, with the most common being nausea at 7% for both doses of CVL-231 compared to 4% with placebo. All other side effects occurred at 2% or less with CVL-231.
Pimavanserin
Pimavanserin, which is approved for the treatment of Parkinson’s psychosis, is currently being studied as an adjunctive treatment to antipsychotics to address negative and residual positive symptoms.56 Pimavanserin is a potent inverse agonist and antagonist at 5-HT2A serotonin receptors and has less potent action at 5-HT2C receptors (see part C of Figure 6). This agent has no direct action at dopamine, adrenergic, histamine, serotonin, or cholinergic receptors.58
The efficacy of pimavanserin 20 mg and 34 mg for treating negative symptoms was evaluated in a 6-month, double-blind, placebo-controlled phase 2B study called the ADVANCE trial.56 After 26 weeks, the patients in the pimavanserin group showed significantly more improvement on the Negative Symptom Assessment-16 (NSA) compared with those receiving placebo, but with only a modest effect size of 0.2. However, at the 34-mg dose approved for Parkinson’s psychosis, the effect size was higher, at 0.34. Interestingly, pimavanserin showed no significant benefit over placebo on the PANSS-Negative symptoms score, the NSA-16 Negative Symptoms subscale score, or the CGI Schizophrenia-Severity scale. Side effects were minimal, with the most frequent being headache (6.5% vs 5% with placebo) and somnolence (5.5% vs 5% with placebo).
A second double-blind, placebo-controlled study called the ENHANCE trial56 investigated pimavanserin as an adjunctive treatment for residual positive symptoms. After 6 weeks of treatment, those receiving adjunctive pimavanserin failed to show significantly greater improvement in either PANSS total or positive symptom scale scores compared with those receiving placebo. However, in a preplanned subgroup analysis of the 80% of the sample that was investigated at non-US sites, pimavanserin did separate from placebo for positive symptoms; in the entire sample, pimavanserin separated from placebo regarding negative symptoms. Pimavanserin was well tolerated in both the ADVANCE and ENHANCE trials. The occurrence of side effects was comparable in the pimavanserin and placebo groups, with the most commonly reported side effects being headache, somnolence, and insomnia (pimavanserin: 5%–7%, placebo: 3%–9%).
Conclusion
In summary, novel mechanisms of action are sorely needed in the treatment of schizophrenia. Previously, lack of knowledge about the pathophysiology of schizophrenia made it difficult to develop and evaluate effective novel mechanisms of action. Advances in imaging technology and drug design have created the potential for new treatments that do not directly block postsynaptic dopamine receptors. This may lead to treatments that not only are effective for patients who do not respond to currently available antipsychotics, but also do not lead to the side effects that stem from dopamine receptor blockade, including parkinsonism, akathisia, prolactin elevation, and weight gain, among others.
Encouraging phase 2 results exist for the TAAR1 agonist, ulotaront, indicating efficacy for total psychotic symptoms with improvement on all PANSS subscale scores. Phase 2B results are available for the M1/M4 agonist, xanomeline, coupled with the peripheral anticholinergic, trospium, with efficacy for total psychotic symptoms and improved subscale scores on the PANSS. Positive phase 1B trial results show efficacy of the M4 positive allosteric modulator, CVL-231, for total symptoms and negative symptoms, and the adjunctive use of the serotonin 2A inverse agonist/antagonist, pimavanserin, for the improvement of negative symptoms. These phase 1B and phase 2B trial results are promising and exciting but must be confirmed with larger phase 3 trials before regulatory approval can be sought.
Any of these mechanisms of action would represent a significant advance in the treatment of schizophrenia. Great hope exists that at least one of these agents with novel mechanisms will be successful in a phase 3 trial so that patients and families can benefit from this new kind of treatment.
Clinical Points
- Schizophrenia symptoms typically emerge in late adolescence or early adulthood, but brain changes underlying the symptoms begin during gestation and continue through the lifespan. Many symptoms are thought to involve dysfunction in neural networks that are essential for processing information and controlling behavior.
- Although more than 20 antipsychotics are available, they are all postsynaptic D2 blockers and share similar benefits and limitations. Discontinuation of antipsychotics may stem from lack of efficacy, intolerable side effects, or worsening of cognitive or negative symptoms.
- Four medications, ulotaront, xanomeline+trospium, CVL-231, and pimavanserin, have shown promise for treating symptoms in schizophrenia without blocking the postsynaptic dopamine receptor.
Published online: February 15, 2022.
Faculty Disclosure
In the spirit of full disclosure, the faculty were asked to complete a statement regarding all relevant personal and financial relationships between themselves or their spouse/partner and any commercial interest. Faculty financial disclosures are as follows:
Dr Abi-Dargham has been a consultant for Neurocrine and Boehringer-Ingelheim; has received honoraria from Sunovion, Otsuka, and Merck; and has received other financial/material support from System1 Biosciences and Terran Biosciences.
Dr Howes has been an employee of Lundbeck; has been a consultant for Angelini, Boehringer-Ingelheim, Eli Lilly, Global Medical Education, Invicro, Janssen, Mylan, Otsuka, Recordati, and Viatris; and has received grant/research support from Autifony, Biogen, Heptares, Neurocrine, Roche, and Sunovion.
Dr Correll has been a consultant for AbbVie, Acadia, Alkermes, Allergan, Angelini, Axsome, Cardio Diagnostics, Gedeon Richter, Holmusk, IntraCellular Therapies, Janssen/J&J, Karuna, LB Pharma, Lundbeck, MedAvante-ProPhase, MedinCell, Medscape, Merck, Mindpax, Mylan, Neurocrine, Noven, Otsuka, Pfizer, Recordati, Relmada, Rovi, Seqirus, Servier, SK Life Science, Sumitomo Dainippon, Sunovion, Supernus, Takeda, Teva, and Viatris; has received grant/research support from Janssen and Takeda; has received honoraria from AbbVie, Acadia, Alkermes, Allergan, Angelini, Aristo, Axsome, Damitsa, Gedeon Richter, Hikma, Holmusk, IntraCellular Therapies, Janssen/J&J, Karuna, LB Pharma, Lundbeck, MedAvante-ProPhase, MedinCell, Medscape, Merck, Mitsubishi Tanabe Pharma, Mylan, Neurocrine, Noven, Otsuka, Pfizer, Recordati, Relmada, Rovi, Seqirus, Servier, SK Life Science, Sumitomo Dainippon, Sunovion, Supernus, Takeda, Teva, and Viatris; has participated in advisory boards for AbbVie, Acadia, Alkermes, Allergan, Angelini, Aristo, Axsome, Damitsa, Gedeon Richter, Hikma, IntraCellular Therapies, Janssen/J&J, Karuna, LB Pharma, Lundbeck, MedAvante-ProPhase, MedinCell, Merck, Mitsubishi Tanabe Pharma, Mylan, Neurocrine, Noven, Otsuka, Recordati, Rovi, Servier, SK Life Science, Sumitomo Dainippon, Sunovion, Supernus, Takeda, Teva, and Viatris; and is a stock shareholder in Cardio Diagnostics, Mindpax, and LB Pharma.
Review Process
The faculty for this InfoPack discussed the content in a series of peer-review planning teleconferences, the chair and faculty reviewed the InfoPack for accuracy, and a member of the Journal Editorial Board who is without conflict of interest reviewed the InfoPack to determine whether the material is evidence-based and objective.
Additional Resources
Summarized article from N ENGL J MED 2020;382(16):1497–1506
See a short video explaining how trace amine–associated receptor 1 (TAAR1) affects neurotransmitters in the brain.
This video is a publicly available educational resource provided by Sunovion Pharmaceuticals Inc. It was produced prior to and independent of the JCP InfoPack “Emerging Treatments in Schizophrenia.”
References (58)

- Koblan KS, Kent J, Hopkins SC, et al. A non-D2-receptor-binding drug for the treatment of schizophrenia. N Engl J Med. 2020;382(16):1497–1506. PubMed CrossRef
- Dedic N, Jones PG, Hopkins SC, et al. SEP-363856, a novel psychotropic agent with a unique, non-D2 receptor mechanism of action. J Pharmacol Exp Ther. 2019;371(1):1–14. PubMed CrossRef
- Correll CU, Koblan KS, Hopkins SC, et al. Safety and effectiveness of ulotaront (SEP-363856) in schizophrenia: results of a 6-month, open-label extension study. NPJ Schizophr. 2021;7(1):63. PubMed CrossRef
- Lieberman JA, Jarskog LF, Malaspina D. Preventing clinical deterioration in the course of schizophrenia: the potential for neuroprotection. J Clin Psychiatry. 2006;67(6):983–990. PubMed CrossRef
- Insel TR. Rethinking schizophrenia. Nature. 2010;468(7321):187–193. PubMed CrossRef
- Ellison-Wright I, Glahn DC, Laird AR, et al. The anatomy of first-episode and chronic schizophrenia: an anatomical likelihood estimation meta-analysis. Am J Psychiatry. 2008;165(8):1015–1023. PubMed CrossRef
- Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28(2):325–334. PubMed CrossRef
- Lener MS, Wong E, Tang CY, et al. White matter abnormalities in schizophrenia and schizotypal personality disorder. Schizophr Bull. 2015;41(1):300–310. PubMed CrossRef
- Schobel SA, Lewandowski NM, Corcoran CM, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66(9):938–946. PubMed CrossRef
- Horga G, Schatz KC, Abi-Dargham A, et al. Deficits in predictive coding underlie hallucinations in schizophrenia. J Neurosci. 2014;34(24):8072–8082. PubMed CrossRef
- Yeo BTT, Krienen FM, Sepulcre J, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol. 2011;106(3):1125–1165. PubMed CrossRef
- Horga G, Cassidy CM, Xu X, et al. Dopamine-related disruption of functional topography of striatal connections in unmedicated patients with schizophrenia. JAMA Psychiatry. 2016;73(8):862–870. PubMed CrossRef
- Surmeier DJ, Ding J, Day M, et al. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30(5):228–235. PubMed CrossRef
- Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr Bull. 2009;35(3):549–562. PubMed CrossRef
- Kegeles LS, Abi-Dargham A, Frankle WG, et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67(3):231–239. PubMed CrossRef
- Howes OD, Montgomery AJ, Asselin MC, et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry. 2009;66(1):13–20. PubMed CrossRef
- Radua J, Schmidt A, Borgwardt S, et al. Ventral striatal activation during reward processing in psychosis: a neurofunctional meta-analysis. JAMA Psychiatry. 2015;72(12):1243–1251. PubMed CrossRef
- Schultz W. Dopamine reward prediction error coding. Dialogues Clin Neurosci. 2016;18(1):23–32. PubMed CrossRef
- Katthagen T, Kaminski J, Heinz A, et al. Striatal dopamine and reward prediction error signaling in unmedicated schizophrenia patients. Schizophr Bull. 2020;46(6):1535–1546. PubMed CrossRef
- Arias-Carrión O, Stamelou M, Murillo-Rodríguez E, et al. Dopaminergic reward system: a short integrative review. Int Arch Med. 2010;3(1):24. PubMed
- O’Donnell P. Adolescent onset of cortical disinhibition in schizophrenia: insights from animal models. Schizophr Bull. 2011;37(3):484–492. PubMed CrossRef
- Slifstein M, van de Giessen E, Van Snellenberg J, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry. 2015;72(4):316–324. PubMed CrossRef
- Cassidy CM, Van Snellenberg JX, Benavides C, et al. Dynamic connectivity between brain networks supports working memory: relationships to dopamine release and schizophrenia. J Neurosci. 2016;36(15):4377–4388. PubMed CrossRef
- Lobb CJ, Wilson CJ, Paladini CA. A dynamic role for GABA receptors on the firing pattern of midbrain dopaminergic neurons. J Neurophysiol. 2010;104(1):403–413. PubMed CrossRef
- McCutcheon RA, Krystal JH, Howes OD. Dopamine and glutamate in schizophrenia: biology, symptoms and treatment. World Psychiatry. 2020;19(1):15–33. PubMed CrossRef
- Driesen NR, McCarthy G, Bhagwagar Z, et al. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol Psychiatry. 2013;18(11):1199–1204. PubMed CrossRef
- Marsman A, van den Heuvel MP, Klomp DWJ, et al. Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophr Bull. 2013;39(1):120–129. PubMed CrossRef
- Raedler TJ, Bymaster FP, Tandon R, et al. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry. 2007;12(3):232–246. PubMed CrossRef
- Scarr E, Gibbons AS, Neo J, et al. Cholinergic connectivity: it’s implications for psychiatric disorders. Front Cell Neurosci. 2013;7:55. PubMed CrossRef
- Rutigliano G, Accorroni A, Zucchi R. The case for TAAR1 as a modulator of central nervous system function. Front Pharmacol. 2018;8:987. PubMed CrossRef
- Berry MD, Gainetdinov RR, Hoener MC, et al. Pharmacology of human trace amine-associated receptors: therapeutic opportunities and challenges. Pharmacol Ther. 2017;180:161–180. PubMed CrossRef
- Espinoza S, Salahpour A, Masri B, et al. Functional interaction between trace amine-associated receptor 1 and dopamine D2 receptor. Mol Pharmacol. 2011;80(3):416–425. PubMed CrossRef
- Dedic N, Dworak H, Zeni C, et al. Therapeutic potential of TAAR1 agonists in schizophrenia: evidence from preclinical models and clinical studies. Int J Mol Sci. 2021;22(24):13185. PubMed CrossRef
- Rutigliano G, Zucchi R. Molecular variants in human trace amine-associated receptors and their implications in mental and metabolic disorders. Cell Mol Neurobiol. 2020;40(2):239–255. PubMed CrossRef
- López-Muñoz F, Alamo C, Cuenca E, et al. History of the discovery and clinical introduction of chlorpromazine. Ann Clin Psychiatry. 2005;17(3):113–135. PubMed CrossRef
- Kaar SJ, Natesan S, McCutcheon R, et al. Antipsychotics: mechanisms underlying clinical response and side-effects and novel treatment approaches based on pathophysiology. Neuropharmacology. 2020;172:107704. PubMed CrossRef
- Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192(4238):481–483. PubMed CrossRef
- Seeman P, Lee T, Chau-Wong M, et al. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261(5562):717–719. PubMed CrossRef
- Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514–520. PubMed CrossRef
- Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927–942. PubMed CrossRef
- Kahn RS, Fleischhacker WW, Boter H, et al; EUFEST study group. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet. 2008;371(9618):1085–1097. PubMed CrossRef
- Stroup TS, Lieberman JA, McEvoy JP, et al; CATIE Investigators. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. Am J Psychiatry. 2006;163(4):611–622. PubMed CrossRef
- Lally J, Ajnakina O, Di Forti M, et al. Two distinct patterns of treatment resistance: clinical predictors of treatment resistance in first-episode schizophrenia spectrum psychoses. Psychol Med. 2016;46(15):3231–3240. PubMed CrossRef
- Demjaha A, Lappin JM, Stahl D, et al. Antipsychotic treatment resistance in first-episode psychosis: prevalence, subtypes and predictors. Psychol Med. 2017;47(11):1981–1989. PubMed CrossRef
- Beck K, McCutcheon R, Stephenson L, et al. Prevalence of treatment-resistant psychoses in the community: a naturalistic study. J Psychopharmacol. 2019;33(10):1248–1253. PubMed CrossRef
- Leung CCY, Gadelrab R, Ntephe CU, et al. Clinical course, neurobiology and therapeutic approaches to treatment resistant schizophrenia. toward an integrated view. Front Psychiatry. 2019;10:601. PubMed CrossRef
- McCutcheon RA, Reis Marques T, Howes OD. Schizophrenia-an overview. JAMA Psychiatry. 2020;77(2):201–210. PubMed CrossRef
- Artaloytia JF, Arango C, Lahti A, et al. Negative signs and symptoms secondary to antipsychotics: a double-blind, randomized trial of a single dose of placebo, haloperidol, and risperidone in healthy volunteers. Am J Psychiatry. 2006;163(3):488–493. PubMed CrossRef
- Kim E, Howes OD, Turkheimer FE, et al. The relationship between antipsychotic D2 occupancy and change in frontal metabolism and working memory: a dual [(11)C]raclopride and [(18) F]FDG imaging study with aripiprazole. Psychopharmacology (Berl). 2013;227(2):221–229. PubMed CrossRef
- Knegtering H, van den Bosch R, Castelein S, et al. Are sexual side effects of prolactin-raising antipsychotics reducible to serum prolactin? Psychoneuroendocrinology. 2008;33(6):711–717. PubMed CrossRef
- Pillinger T, McCutcheon RA, Vano L, et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: a systematic review and network meta-analysis. Lancet Psychiatry. 2020;7(1):64–77. PubMed CrossRef
- Miller GM. The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity. J Neurochem. 2011;116(2):164–176. PubMed CrossRef
- Kokkinou M, Irvine EE, Bonsall DR, et al. Reproducing the dopamine pathophysiology of schizophrenia and approaches to ameliorate it: a translational imaging study with ketamine. Mol Psychiatry. 2021;26(6):2562–2576. PubMed CrossRef
- Brannan SK, Sawchak S, Miller AC, et al. Muscarinic cholinergic receptor agonist and peripheral antagonist for schizophrenia. N Engl J Med. 2021;384(8):717–726. PubMed CrossRef
- Transforming the Possible in Neuroscience: Topline Data for Phase 1b Trial of CVL-231 in Schizophrenia. Cerevel website. https://investors.cerevel.com/static-files/31d256be-15f7-4340-8af3-7cdd058308fe. Published June 2021.
- Davis J, Zamora D, Horowitz M, et al. Evaluating pimavanserin as a treatment for psychiatric disorders: a pharmacological property in search of an indication. Expert Opin Pharmacother. 2021;22(13):1651–1660. PubMed CrossRef
- Cerevel Therapeutics Announces Positive Topline Results for CVL-231 in Phase 1b Clinical Trial in Patients with Schizophrenia. Cerevel website. https://investors.cerevel.com/news-releases/news-release-details/cerevel-therapeutics-announces-positive-topline-results-cvl-231/. Published June 29, 2021. Accessed December 29, 2021.
- Hunter NS, Anderson KC, Cox A. Pimavanserin. Drugs Today (Barc). 2015;51(11):645–652. PubMed CrossRef
This PDF is free for all visitors!


