Objective: To assess putative antidepressant and procognitive effects of decoglurant, a selective metabotropic glutamate receptor type 2/3 (mGlu2/3) negative allosteric modulator, as adjunctive treatment to selective serotonin reuptake inhibitors and/or serotonin-norepinephrine reuptake inhibitors (SSRIs/SNRIs) in patients with partially refractory major depressive disorder (MDD), diagnosed using DSM-IV-TR criteria.
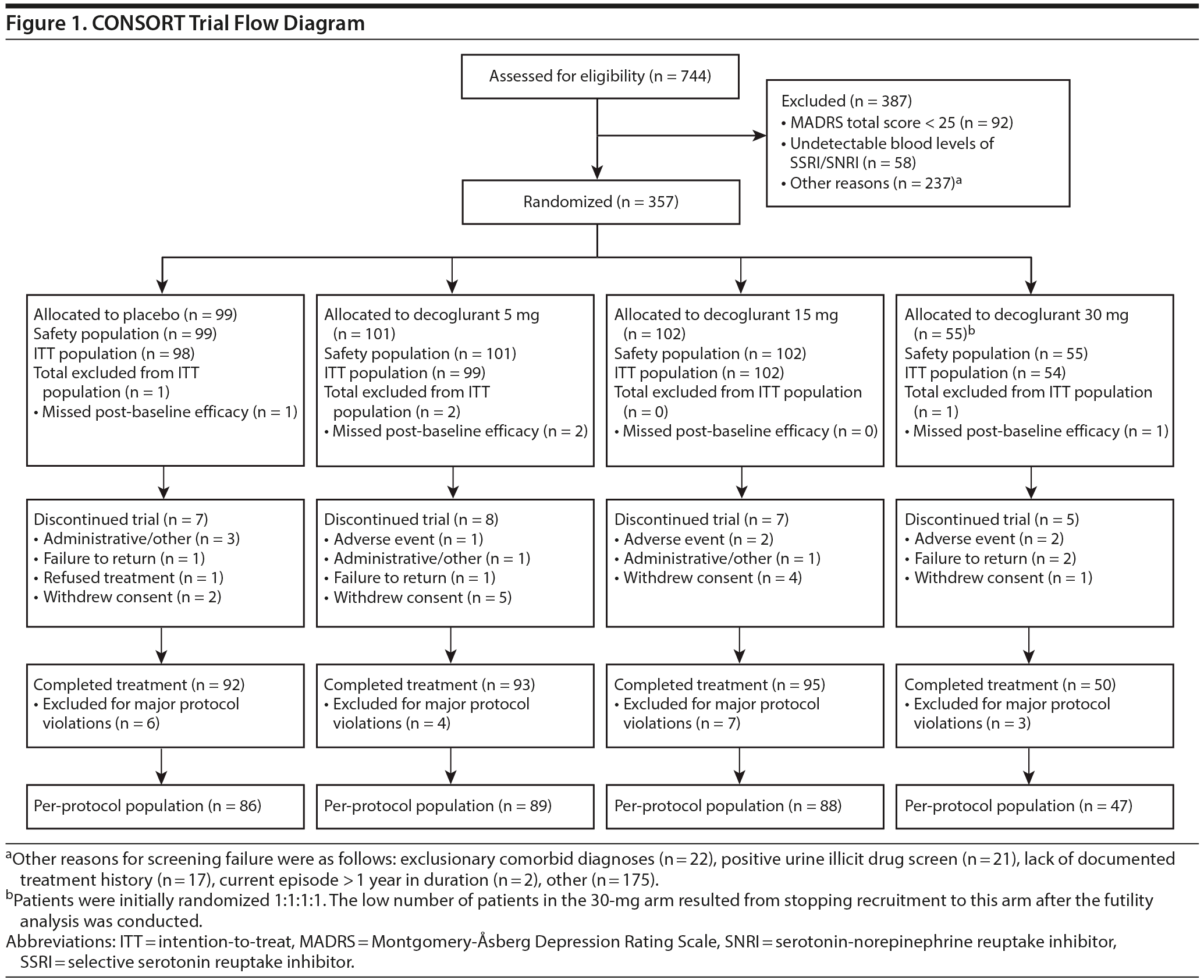
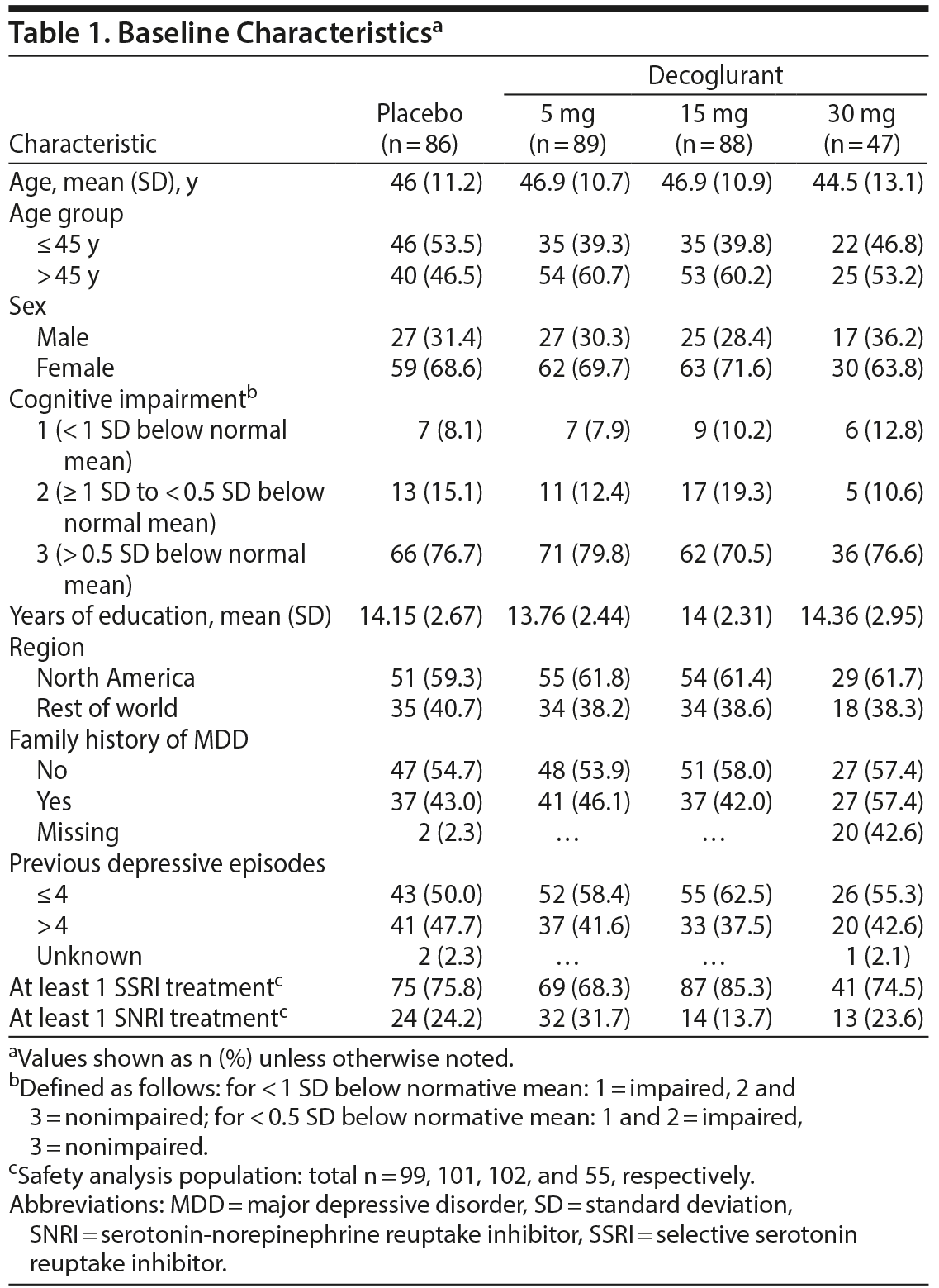
Methods: This randomized, placebo-controlled, double-blind, multicenter phase 2 trial consisted of 4 weeks’ screening, 6 weeks’ treatment, and 8 weeks’ follow-up between September 2011 and June 2014. Individuals with Montgomery-Åsberg Depression Rating Scale (MADRS) score ≥ 25 and Clinical Global Impressions-Severity of Illness scale score ≥ 4, despite up to 2 adequate trials of an SSRI/SNRI and compliance confirmed by positive SSRI/SNRI blood levels, were randomized to decoglurant 5 mg (n = 101), 15 mg (n = 102), or 30 mg (n = 55) daily or placebo (n = 99) as adjunct to ongoing treatment with 1 SSRI/SNRI. An adaptive design was used with an interim analysis after 30 patients in each group had received 6 weeks’ treatment. The primary outcome variable was change in MADRS total score from baseline to end of treatment. Primary assessments were performed by fully blinded centralized raters.
Results: Of 357 participants, 310 completed 6 weeks’ treatment. At 6 weeks, no significant differences between any active treatment arm and placebo in reducing MADRS total score or response or remission rates were observed. Decoglurant exerted no significant effects on Cambridge Neuropsychological Test Automated Battery cognitive accuracy and cognitive speed composite scores or on secondary measures of mood and functioning. A relatively high placebo response was observed, which may have constrained the ability to detect treatment effects. No deaths occurred; few patients reported serious adverse events.
Conclusions: Decoglurant was well tolerated overall but did not exert any antidepressant or procognitive effects.
Trial Registration: ClinicalTrials.gov identifier: NCT01457677
This work may not be copied, distributed, displayed, published, reproduced, transmitted, modified, posted, sold, licensed, or used for commercial purposes. By downloading this file, you are agreeing to the publisher’s Terms & Conditions.
ABSTRACT
Objective: To assess putative antidepressant and procognitive effects of decoglurant, a selective metabotropic glutamate receptor type 2/3 (mGlu2/3) negative allosteric modulator, as adjunctive treatment to selective serotonin reuptake inhibitors and/or serotonin-norepinephrine reuptake inhibitors (SSRIs/SNRIs) in patients with partially refractory major depressive disorder (MDD), diagnosed using DSM-IV-TR criteria.
Methods: This randomized, placebo-controlled, double-blind, multicenter phase 2 trial consisted of 4 weeks’ screening, 6 weeks’ treatment, and 8 weeks’ follow-up between September 2011 and June 2014. Individuals with Montgomery-Åsberg Depression Rating Scale (MADRS) score ≥ 25 and Clinical Global Impressions-Severity of Illness scale score ≥ 4, despite up to 2 adequate trials of an SSRI/SNRI and compliance confirmed by positive SSRI/SNRI blood levels, were randomized to decoglurant 5 mg (n = 101), 15 mg (n = 102), or 30 mg (n = 55) daily or placebo (n = 99) as adjunct to ongoing treatment with 1 SSRI/SNRI. An adaptive design was used with an interim analysis after 30 patients in each group had received 6 weeks’ treatment. The primary outcome variable was change in MADRS total score from baseline to end of treatment. Primary assessments were performed by fully blinded centralized raters.
Results: Of 357 participants, 310 completed 6 weeks’ treatment. At 6 weeks, no significant differences between any active treatment arm and placebo in reducing MADRS total score or response or remission rates were observed. Decoglurant exerted no significant effects on Cambridge Neuropsychological Test Automated Battery cognitive accuracy and cognitive speed composite scores or on secondary measures of mood and functioning. A relatively high placebo response was observed, which may have constrained the ability to detect treatment effects. No deaths occurred; few patients reported serious adverse events.
Conclusions: Decoglurant was well tolerated overall but did not exert any antidepressant or procognitive effects.
Trial Registration: ClinicalTrials.gov identifier: NCT01457677
J Clin Psychiatry 2020;81(4):18m12470
To cite: Umbricht D, Niggli M, Sanwald-Ducray P, et al. Randomized, double-blind, placebo-controlled trial of the mGlu2/3 negative allosteric modulator decoglurant in partially refractory major depressive disorder. J Clin Psychiatry. 2020;81(4):18m12470.
To share: https://doi.org/10.4088/JCP.18m12470
© Copyright 2020 Physicians Postgraduate Press, Inc.
aRoche Pharmaceutical Research and Early Development, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd, Basel, Switzerland
bRoche Product Development Biometrics, F. Hoffmann-La Roche Ltd, Basel, Switzerland
cRoche Pharmaceutical Research and Early Development, Roche Innovation Center New York, F. Hoffmann-La Roche Ltd, New York, New York
dRoche Product Development Neuroscience, F. Hoffmann-La Roche Ltd, Basel, Switzerland
*Corresponding author: Daniel Umbricht, MD, Roche Pharmaceutical Research and Early Development, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd, Bldg 1/Room 16.N655, Grenzacherstrasse 124, 4070 Basel, Switzerland ([email protected]).
Major depressive disorder (MDD) exerts a significant personal, economic, and societal burden.1-3 Pharmacologic intervention currently involves treatment with monoamine reuptake inhibitors (eg, selective serotonin reuptake inhibitors [SSRIs], serotonin-norepinephrine reuptake inhibitors [SNRIs]), and atypical antipsychotics, in conjunction with psychotherapy. However, a substantial proportion of patients do not respond to first- or second-line treatment.4
The clinical presentation of MDD includes impairments in cognitive functions, which, contrary to previous beliefs, represent a key feature of MDD that is largely independent of the severity of classical depressive symptoms.5-8 Most available antidepressants—with the possible exception of vortioxetine9—have not been shown to improve cognitive deficits beyond those accounted for by depressive symptoms. Novel antidepressants that also effectively ameliorate cognitive deficits associated with MDD are needed.
While classic antidepressants mediate their effect primarily through monoamines, growing evidence supports the treatment of MDD through modulation of dysregulated glutamate neurotransmission.10-13 Of specific interest, as therapeutic targets in MDD, are the metabotropic glutamate receptor type 2 (mGlu2) receptors—presynaptic auto-inhibitory receptors that are highly expressed in the PFC, hippocampus, amygdala, and nucleus accumbens—with preclinical research suggesting that mGlu2 antagonists have antidepressant and procognitive effects.10,14-17 Indeed, a highly selective mGlu2/3 antagonist (RO4432717) that was used as a tool compound reversed mGlu2/3 agonist induced cognitive deficits and improved long-term memory in preclinical assays.5 Another selective, potent non-competitive mGlu2/3 negative allosteric modulator, decoglurant (RG1578),18 was found to reduce the anhedonia index in a chronic mild stress model in rats and to rescue scopolamine-induced deficits in executive function and attention in non-human primates (D.U., unpublished data, 2015). We therefore hypothesized that in individuals diagnosed with MDD, decoglurant may restore normal glutamate transmission, thus reducing depressive and cognitive symptoms.
In initial safety studies in healthy volunteers, decoglurant had a good safety and tolerability profile (EudraCT trial no. 2009-011624-62). A potential procognitive effect was also supported by its reversal of scopolamine-induced cognitive deficits in healthy volunteers (EudraCT trial no. 2009-014678-17).
The primary objective of the current trial was to assess putative antidepressant and procognitive effects of decoglurant versus placebo as adjunctive treatment to SSRI/SNRI therapy in individuals with MDD and inadequate response to antidepressant treatment. The aim was to focus on patients considered to have an optimum chance of responding to a novel adjunctive treatment option, specifically those with relatively recent-onset disease failing no more than 2 previous treatments.
METHODS
Trial Design and Patients
This randomized, placebo-controlled, double-blind, phase 2 trial consisted of a 4-week screening period, 6-week treatment period, and 8-week follow-up period (ClinicalTrials.gov identifier: NCT01457677). It was conducted between September 2011 and June 2014 at 72 sites in Canada, Austria, Germany, Russia, Ukraine, Slovakia, South Africa, and the United States following Guidelines for Good Clinical Practice.19 The protocol was approved by the health authorities of each country and ethics committees of each site. All participants gave written informed consent.
Patients with a Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR), diagnosis of MDD without psychotic features, who continued to have depressive symptoms despite 1 or 2 adequate trials with an SSRI or SNRI at doses equal to or greater than the accepted dose according to the Massachusetts General Hospital Antidepressant Treatment Response Questionnaire,20 were eligible. The index depressive episode had to have started within 1 year of screening, and treatment dose and duration were verified from the treating physician and/or pharmacy records. Other key inclusion criteria were scores of ≥ 25 on the Montgomery-Åsberg Depression Rating Scale (MADRS)21 and ≥ 4 on the Clinical Global Impressions-Severity of Illness scale (CGI-S),22 as assessed by fully blinded centralized raters. Compliance with current SSRI/SNRI treatment was assessed with blood tests; if levels of the respective antidepressant were undetectable, patients were not enrolled. Only 1 SSRI/SNRI was continued during the trial. Medications used to treat stable medical conditions other than depression were allowed, providing doses were stable (see Supplementary Table 1 for full inclusion and exclusion criteria).
- Many patients with major depressive disorder (MDD) do not respond to first- or second-line therapies, and current treatments do not improve the associated cognitive deficits.
- In patients with partially treatment-resistant MDD, the metabotropic glutamate receptor type 2/3 (mGlu2/3) antagonist decoglurant was well tolerated but did not have an antidepressant or procognitive effects in combination with selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors.
Randomization
After screening, participants were randomized into 4 groups: decoglurant at a once-daily dose of 5 mg, 15 mg, or 30 mg or placebo, all in addition to existing permitted medications (see Supplementary Table 2 for details on the administration of study medication).
Randomization codes generated by the sponsor were administered by an interactive voice or web-based response system. Randomization was stratified by cognitive impairment, sex, and geographic region. Cognitive impairment was initially defined as a score of 1 standard deviation below the normative mean of the mean combined score of the attentional (Rapid Visual Processing [RVP]), memory (Paired Associates Learning [PAL]), and executive (Stockings of Cambridge [SOC]) tests of the Cambridge Neuropsychological Test Automated Battery (CANTAB).23 After recruitment of 79 participants, the threshold was lowered to 0.5 SD below the normative mean as too few patients met the original criterion. Before this change, 13 patients were categorized as “not cognitively impaired”; subsequently, they would have been categorized as “cognitively impaired.”
Endpoints
The primary efficacy endpoint was the MADRS total score, assessed by centralized, fully blinded raters using the MADRS-SIGMA revision.24
Secondary mood and functioning endpoints included the CGI-S and CGI-Improvement scale (CGI-I)22 and participant-rated measures comprising the Inventory of Depressive Symptomatology-Self-Report 30-item version (IDS-SR30),25 Cognitive and Physical Functioning Questionnaire (CPFQ),26 Sheehan Disability Scale (SDS),27 Quality of Life Enjoyment and Satisfaction Questionnaire-Short Form (Q-LES-Q-SF),28 and Patient-Rated Global Improvement (PGI), adapted from the clinician-rated CGI-S.22
Cognitive impairment was assessed with the CANTAB cognitive test battery,23 which included Motor Screening (MOT); RVP; Delayed Matched to Sample (DMS); Emotional Recognition Task (ERT); PAL; SOC at screening, or One-Touch Stockings of Cambridge (OTS, at baseline and day 42); and Attention Shifting Test (AST). A factor analysis—using 2 key parameters for each test of the CANTAB battery (excluding the ERT)—of the data obtained at baseline in the current study, and a simultaneously conducted study of basimglurant in treatment-refractory MDD,13 demonstrated two key factors: one loading on accuracy measures of all tests and one loading on measures of reaction time across all tests (see Supplementary Table 3 for exploratory factor analysis). Thus, an a priori decision was made before study completion to use calculated cognitive accuracy and speed composite scores as the primary cognitive outcome variables (see Supplementary Table 4 for details of the tasks tested for CANTAB).
Safety data were collected through clinical and neurologic examinations; recording of adverse events (AEs); clinician-administered rating scales (Columbia-Suicide Severity Rating Scale,29 Extrapyramidal Symptom Rating Scale-Abbreviated,30 and Young Mania Rating Scale [item 1 when indicated to follow up on AEs only]31), and measurement of vital signs, electrocardiograms, and laboratory parameters.
Assessments
Centralized raters assessed illness severity at screening using the MADRS, and they administered the MADRS and CGI-S at baseline and at all visits during treatment and follow-up periods via telephone in the patient’s native language. Site raters (a local trial-site physician, nurse, clinical psychologist, or social worker with certified psychiatric practice and ≥ 2 years’ experience administering standardized rating scales in MDD) also administered the MADRS and CGI-S at baseline and week 6 and the CGI-I at week 6 (see Supplementary Figure 1 for details of the trial design).
At screening, assessments included the centrally administered MADRS and CGI-S, the patient-rated IDS-SR30, and the CANTAB battery. Primary and secondary outcome assessments were conducted at baseline, weekly during the 6-week treatment period, and at weeks 8, 10, and 14 during the follow-up period (Supplementary Figure 1). The CANTAB battery was administered at screening, baseline, and week 6. A limited CANTAB battery including only the MOT, RVP, and PAL was administered at day 7.
Statistical Analysis
The primary efficacy variable—change in MADRS total score from baseline to end of treatment—was analyzed using a mixed-effects model for repeated measures (MMRM) that included independent variables of the fixed effects of treatment, stratification variables, geographical region, assessment weeks relative to the first dose of study medication (ie, time), and treatment-by-time interaction, along with the continuous effect of baseline MADRS total score. An unstructured variance-covariance matrix was applied to model the within-patient errors. A treatment-by-time interaction contrast was used to estimate the difference between each decoglurant dose and placebo in mean change from baseline to week 6 of treatment. Response was defined as ≥ 50% improvement from baseline in MADRS total score and remission as MADRS total score ≤ 10. The primary analysis was conducted for the per-protocol (PP) population (ie, all randomized patients with valid baseline and 6-week MADRS total scores [centralized rating] who were not excluded because of protocol violation criteria) with no imputation for missing values. The 95% confidence intervals (CIs) of treatment difference and nominal P value (no adjustment for multiple comparisons) are reported for each dose of decoglurant. As a sensitivity analysis, the primary efficacy variable was also analyzed using the intention-to-treat (ITT) population (all patients randomly assigned to treatment) via MMRM or last observation carried forward imputation. A “MADRS interest-activity” score32 was calculated by summing the scores of the Concentration Difficulties, Lassitude, and Inability to Feel items.
Analyses of variance and covariance were used to investigate the effect of decoglurant on the secondary efficacy endpoints using the PP and ITT populations. Subgroup analyses based on categorical variables were performed on selected secondary efficacy endpoints. Ordered categorical data were analyzed using the Wilcoxon signed rank test and binary data using the Fisher exact test.
To minimize exposure to potentially ineffective treatment, and associated side effects, a prespecified Bayesian interim futility analysis was conducted after 30 participants in each treatment arm had completed 6 weeks’ treatment. It was decided before the start of the study to stop 1 dose arm if the probability of reaching an effect size of at least 0.25 between a dose arm and placebo in the primary efficacy measure at the completion of the study was < 20%, taking into account both probability of success and tolerability if 2 or 3 arms met this criterion.
A sample size of 85 evaluable participants per non-dropped treatment arm provided approximately 80% power at a 1-sided α level of 5%, with an effect size of 0.38.
Safety was assessed in all patients who received at least 1 dose of study medication with a post-dose safety assessment (safety population).
RESULTS
Patient Disposition and Baseline Characteristics
A total of 744 individuals were screened; among the screening failures (n = 387), 24% of patients (n = 92) were ineligible owing to MADRS scores below 25 and 15% (n = 58) for absence of detectable levels of antidepressant drug. A total of 357 patients (48% of all screened patients) passed screening and were randomized, and 310 participants completed 6 weeks of treatment without major protocol violations (Figure 1). Major protocol violations were observed in 20 patients, with similar proportions in each treatment arm. The majority of protocol violations (82%) were instances in which less than 80% (< 34) or more than 120% (> 58) of doses were received. Demographic characteristics and antidepressant treatment were well matched across arms (Table 1).
Click figure to enlarge
Click figure to enlarge
Interim Analysis
The interim analysis showed that the Bayesian predictive probabilities of reaching a final effect size of ≥ 0.25 at the end of the study were 6.5%, 3.7%, and 6.6% for decoglurant 5 mg, 15 mg, and 30 mg, respectively. The decoglurant 30-mg arm, which was associated with the highest rate of AEs, was discontinued.
Primary Endpoint
At baseline, the mean (SD) MADRS total score across all treatment groups was 31 (6). At 6 weeks, large decreases in the MADRS total score were observed, but they did not differ significantly between decoglurant treatment and placebo groups (Figure 2, Table 2). Response and remission rates at week 6 (35%-47% and 29%-38% of patients, respectively) did not differ significantly between treatment and placebo groups (see Supplementary Figure 2 for MADRS response and remission rates). The supporting analysis using the ITT population confirmed these findings (Table 2).
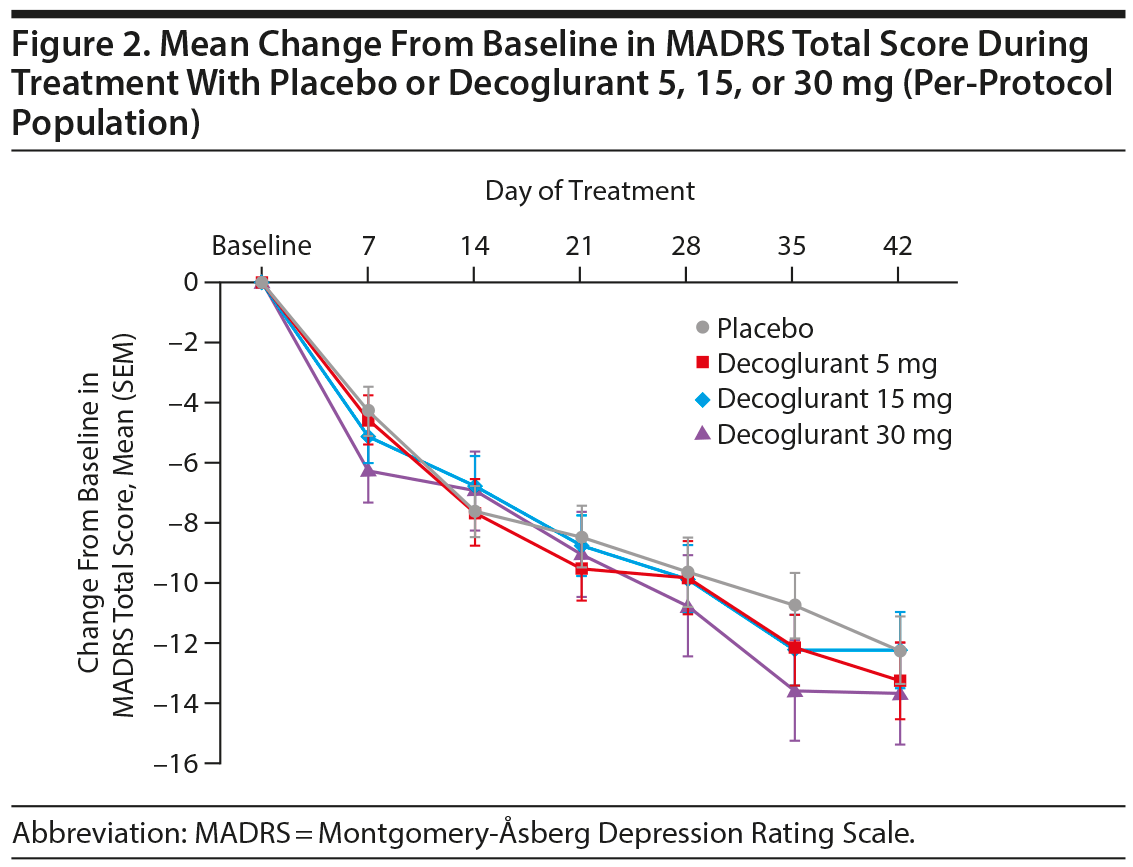
Click figure to enlarge
No significant reductions were observed in interest-activity scores in the treatment groups compared with placebo (data not shown).
Additional analyses in subgroups of participants stratified by sex, age, history of at least 4 previous depressive episodes, a family history of MDD, baseline cognitive impairment, subjective cognitive complaints (CPFQ total score ≥ 25), and geographic region demonstrated no significant treatment effects compared with placebo.
Secondary Endpoints
Mood and functioning. Statistical analyses in both PP and ITT populations demonstrated no significant differences in secondary outcomes between decoglurant and placebo groups. Results for the IDS-SR30 and CPFQ are shown in Table 2 (data for other measures not shown).
Cognitive impairment. At baseline, 24% of participants were categorized as showing cognitive impairment, while the mean cognitive performance of participants was within the normal range. At 6 weeks, none of the decoglurant doses exerted any significant effect on the CANTAB cognitive accuracy and cognitive speed composite scores compared to placebo (Table 2). Similarly, decoglurant did not significantly affect the performance of any of the individual CANTAB tasks in any arm (data not shown). Analyses in patients categorized as cognitively impaired at baseline, and in patients with a performance score of ≤ 85% on the DMS task of CANTAB (as an alternative post hoc definition of cognitive impairment), showed no significant treatment effects compared with placebo (data not shown).
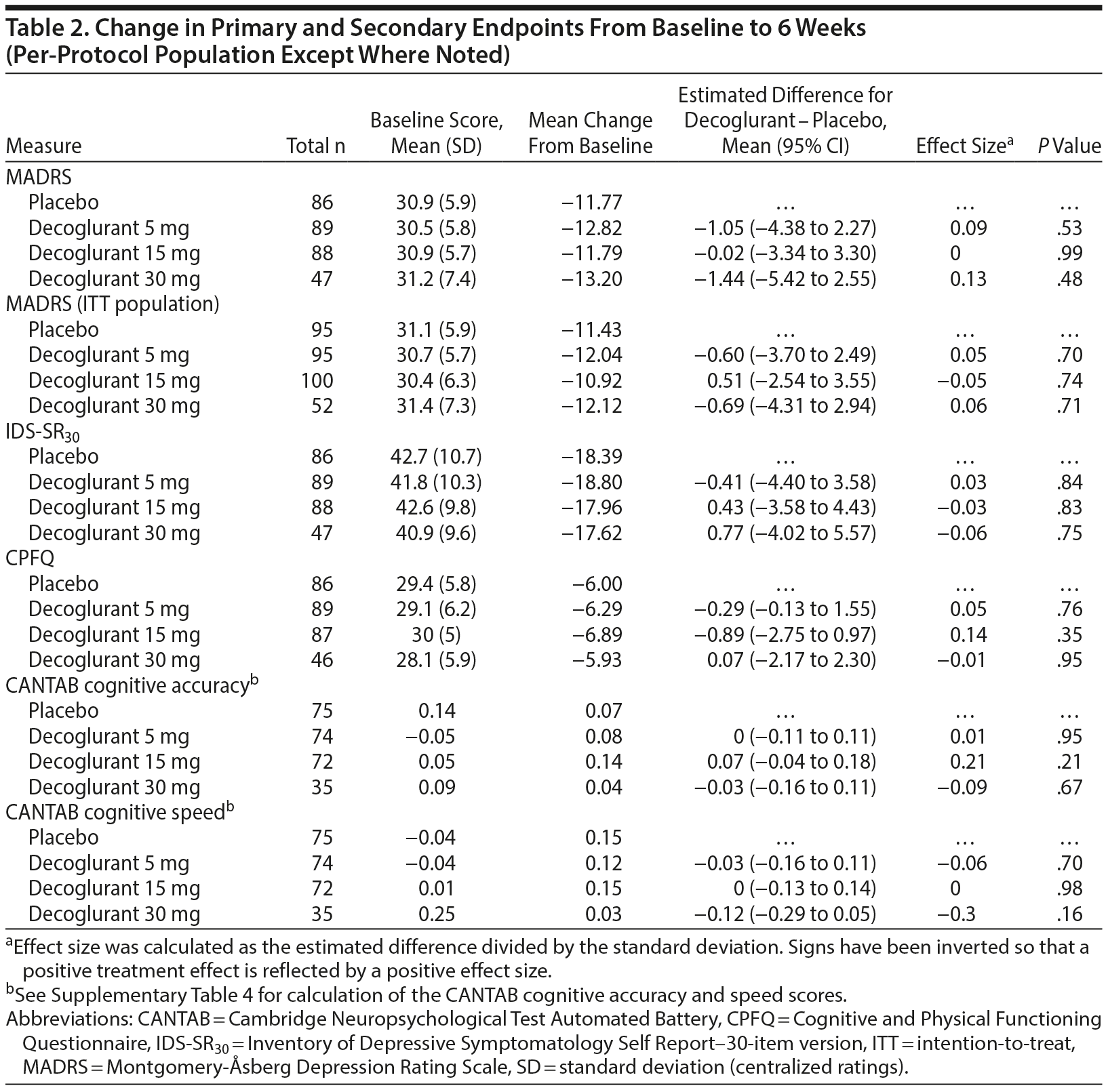
Click figure to enlarge
Centralized Versus Site Raters
Mean changes from baseline in MADRS score as assessed by the centralized raters tended to be smaller than those assessed by site raters, particularly in the placebo group (see Supplementary Table 5 for mean changes from baseline). Response rates were higher based on site assessments, particularly in the placebo group. However, remission rates in the placebo group as assessed by centralized and site raters were comparable (Supplementary Table 5).
Pharmacokinetics and Exposure to Treatment
At 6 weeks, the mean (range) maximum plasma concentration was 143 (1-697) ng/mL with decoglurant 5 mg, 532 (1-2,090) ng/mL with 15 mg, and 835 (8-2,960) ng/mL with 30 mg; exposure thus exceeded the putative minimum therapeutic exposure of 90-100 ng/mL in all arms. Participants were grouped by their mean pre-dose plasma drug concentrations measured weekly during the last 3 weeks of treatment: < 100 ng/mL; ≥ 100-< 200 ng/mL; ≥ 200-< 300 ng/mL; and ≥ 300 ng/mL. No significant effects between exposure group and the primary and secondary endpoints were observed (data not shown). Most participants (85.5%-87.1%) in the 3 treatment arms were treated with decoglurant for > 35 days. The median (range) total dose was 210 (30-220) mg in the 5-mg arm, 630 (60-660) mg in the 15-mg arm, and 1,260 (30-1,320) mg in the 30-mg arm.
Safety
There was a low incidence of trial discontinuation due to AEs (Figure 1). The incidence of any AEs was higher in the decoglurant 30-mg treatment arm (85.5%) compared with the 5-mg (75.2%) and 15-mg (77.5%) treatment arms. The most frequent AEs included headache, nausea, and dizziness (Table 3). No deaths occurred during the trial, and few patients reported serious AEs (Table 3).
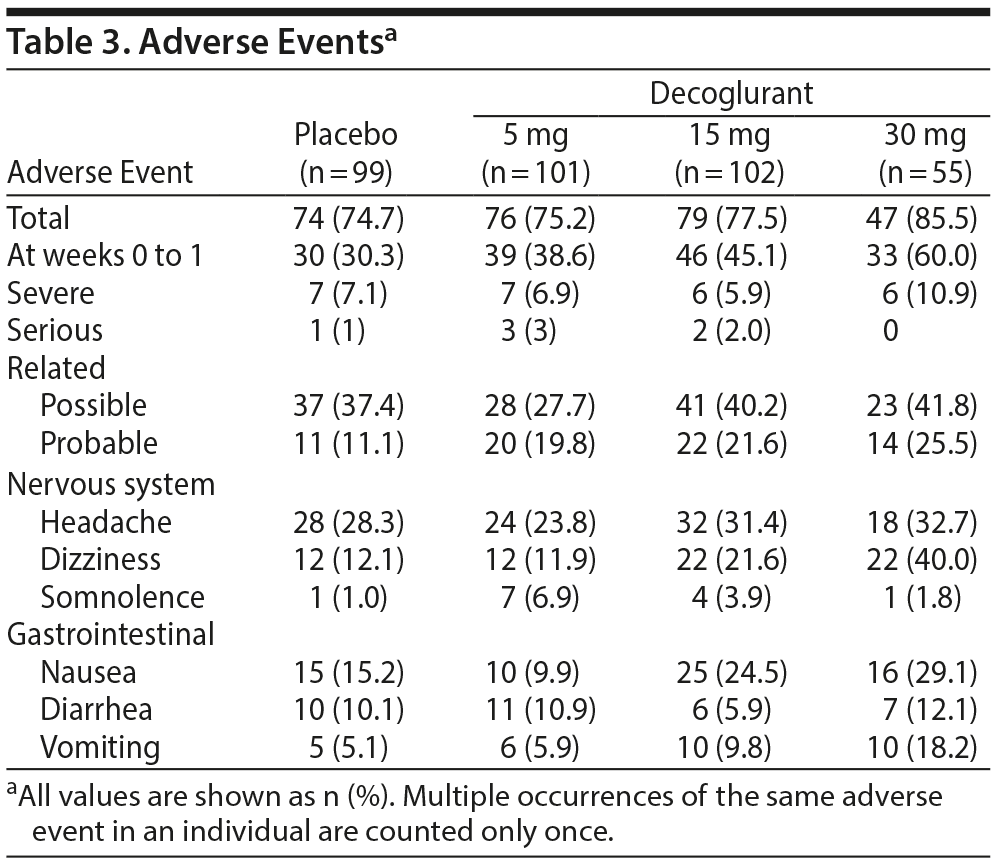
Click figure to enlarge
DISCUSSION
This phase 2 clinical trial investigated putative antidepressant and procognitive effects of the mGlu2/3 antagonist decoglurant compared with placebo in individuals with partially treatment-resistant MDD. As an adjunct to SRRI/SNRI therapy, decoglurant did not demonstrate significant antidepressant or procognitive effects versus placebo and was well tolerated overall.
Drug exposure was adequate in all arms and exceeded the IC80 (90-100 ng/mL). No relationship between mean plasma decoglurant concentration and any main endpoint was observed. In the absence of data confirming target engagement/receptor occupancy in humans, it cannot be ruled out that central exposure to the drug was suboptimal. However, given the high brain penetrance observed in preclinical studies and the central nervous system nature of adverse effects observed in the safety studies in healthy volunteers, suboptimal exposure is highly unlikely.
No significant improvement in performance on the CANTAB cognitive battery was observed with decoglurant. However, the power of the study to detect a treatment effect was limited because of the lower-than-expected prevalence of clinically relevant cognitive impairment. As the primary goal of this study was to demonstrate antidepressant effects, no cognitive impairment threshold was defined as an inclusion criterion. Rather, patients were stratified into those with and those without cognitive impairment. The criterion for cognitive impairment was initially defined as 1 SD below CANTAB normative means, which was reduced to 0.5 SD because of the low number of patients meeting this initial threshold. The unexpected absence of significant cognitive impairment may have arisen because studies reporting clinically relevant cognitive impairment in MDD were performed primarily at academic centers; hence, they may have recruited from different patient populations than commercially oriented research centers. However, the absence of cognitive impairment may have been spurious and resulting from the use of normative data obtained in a restricted population, namely UK residents. Indeed, during this trial, we obtained cognitive data in age-, sex-, and education-matched controls at selected study centers in each country. Compared with these healthy volunteers, our patients were more impaired.
Lastly, the cognitive battery used may not have been optimal to detect procognitive effects of our compound—a hypothesis that cannot be evaluated with this study alone. To detect treatment effects, future clinical trials may need to utilize an explicit cognitive impairment criterion and more than one cognitive battery, along with analyzing cognitive impairment as a primary endpoint.
Clinical trials of antidepressants in MDD have been confounded by high placebo response rates,33-37 although data from large US Food and Drug Administration trials suggest the effect may be less than originally reported.38 To minimize the placebo response, the current trial implemented inclusion criteria (at least moderate MADRS scores and evidence of compliance with the relevant SSRI/SNRI) and also—given the tendency for inflated site-based scoring39—used fully blinded, centralized raters to assess baseline severity and outcome.37 Notably, among the individuals screened, 39% (150 patients) failed these screening criteria because of low MADRS scores or negative blood tests for SSRIs/SNRIs. Despite these initial safeguards, the placebo response and remission rates observed were high relative to previous antidepressant trials: 35% of the placebo group met the MADRS criteria for treatment response, and 29% met the MADRS criteria for full remission of MDD. These results suggest that initial patient selection may be a factor determining placebo response more strongly than outcome assessments performed by raters blinded to the protocol and study visit. We recognize that while the severity of MDD was assessed centrally in our trial, the original diagnosis of MDD was made at the individual sites. Around 15% of patients recruited to trials of MDD and resistant disease may actually be ineligible, mainly on the grounds of inadequate treatment resistance, distorting treatment effects.40 This finding appears to be more pronounced with recruitment at non-academic, as opposed to academic, centers. As we selected patients in whom the duration of the current episode did not exceed 1 year, it can be argued that this selection criterion excluded truly treatment-resistant patients; indeed, the reasoning behind this criterion was to exclude patients in whom the likelihood of any response was expected to be very low. However, our results suggest that the patients entering our study showed, if anything, a very high response to any intervention. Other possible reasons for the high placebo response in our trial include the number of study treatment arms (leading to a probability of only 25% for a patient to be randomized to placebo)37,41 and extended interaction with patients,36,42 both of which have been shown to be positively associated with this effect. The challenge in first-in-patient studies is the selection of the optimal dose. Without any data on target engagement, as in the case of decoglurant, a design with only one active arm is risky, and the researcher is therefore left with a design that may increase placebo response.
Comparison of the placebo response rate to rates in studies with additional safeguards for patient selection suggests that improvements might be achieved by blinded, independent diagnosis of treatment-resistant MDD and assessment of the appropriateness of adjunctive treatment to SSRI/SNRI therapy as well as prospective testing of treatment nonresponse to background therapy before administration of study drug.40,43-45 A double- or single-blind placebo run-in period may also be beneficial.36 Nonetheless, while it is clear that the aforementioned improvements should be implemented for future trials, the absence of any signal of a treatment effect in our study across multiple endpoints in the subgroup analyses supports our conclusion that decoglurant does not exert any antidepressant effects.
To conclude, decoglurant was well tolerated, and, at the doses investigated, no antidepressant or procognitive effects were observed in individuals with partially treatment-resistant MDD compared with placebo. A relatively high placebo response and a seeming absence of clinically relevant cognitive impairment in the majority of patients may have reduced the possibility of demonstrating antidepressant and procognitive effects. An even more careful selection of patients may help to address these issues in future trials.
Submitted: July 17, 2018; accepted May 13, 2020.
Published online: July 14, 2020.
Author contributions: All authors reviewed and revised the manuscript and approved the final version. In addition, Dr Umbricht, Dr Boak, Dr Niggli, and Dr Sanwald-Ducray designed the study and led its implementation, were involved in the acquisition of data, played an important role in interpreting the results, and drafted versions of the manuscript; Dr Deptula, Ms Moore, and Dr Grünbauer were involved in the acquisition of data and played an important role in interpreting the results; and Dr Fontoura was involved in designing the study and played an important role in interpreting the results.
Potential conflicts of interest: All authors were employees of, and owned shares in, F. Hoffmann-La Roche Ltd at the time of this study. Dr Deptula is currently acting as a consultant to Immunobrain Checkpoint Inc.
Funding/support: This study was funded by F. Hoffman-La Roche Ltd.
Role of the sponsor: The sponsor was involved in the design of this study and the collection, analysis, and interpretation of study data. All authors were employees of the sponsor at the time of the study. The development of, and final decision to submit, this manuscript for publication was the responsibility of the authors.
Previous presentation: Results from this study were presented in part as oral presentations at the 70th Annual Meeting of the Society of Biological Psychiatry; May 14-16, 2015; Toronto, Ontario, Canada ▪ the Annual Meeting of the American Society of Clinical Psychopharmacology; June 21-25, 2015; Miami, Florida ▪ the 9th International Meeting on Metabotropic Glutamate Receptors; October 1-6, 2017; Taormina, Italy, and as poster presentations at the 28th ECNP Congress; August 29-September 01, 2015; Amsterdam, The Netherlands ▪ the 54th Annual Meeting of the American College of Neuropsychopharmacology; December 6-10, 2015; Hollywood, Florida.
Acknowledgments: Medical writing assistance, funded by F. Hoffman-La Roche Ltd, was provided by Grace Townshend, MSc, and Charlie Hunt, PhD, of Watermeadow Medical (UK), an Ashfield company. Ms Townshend and Dr Hunt have no conflicts of interest to declare.
Additional information: All reasonable requests for access to study data will be facilitated whenever possible and should be directed to the corresponding author.
Supplementary material: See accompanying pages.
REFERENCES
1.Fostick L, Silberman A, Beckman M, et al. The economic impact of depression: resistance or severity? Eur Neuropsychopharmacol. 2010;20(10):671-675. PubMed CrossRef
2.Salomon JA, Vos T, Hogan DR, et al. Common values in assessing health outcomes from disease and injury: disability weights measurement study for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2129-2143. PubMed CrossRef
3.Lasalvia A, Zoppei S, Van Bortel T, et al; ASPEN/INDIGO Study Group. Global pattern of experienced and anticipated discrimination reported by people with major depressive disorder: a cross-sectional survey. Lancet. 2013;381(9860):55-62. PubMed CrossRef
4.Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917. PubMed CrossRef
5.Goeldner C, Ballard TM, Knoflach F, et al. Cognitive impairment in major depression and the mGlu2 receptor as a therapeutic target. Neuropharmacology. 2013;64:337-346. PubMed CrossRef
6.Bhalla RK, Butters MA, Mulsant BH, et al. Persistence of neuropsychologic deficits in the remitted state of late-life depression. Am J Geriatr Psychiatry. 2006;14(5):419-427. PubMed CrossRef
7.McDermott LM, Ebmeier KP. A meta-analysis of depression severity and cognitive function. J Affect Disord. 2009;119(1-3):1-8. PubMed CrossRef
8.Rock PL, Roiser JP, Riedel WJ, et al. Cognitive impairment in depression: a systematic review and meta-analysis. Psychol Med. 2014;44(10):2029-2040. PubMed CrossRef
9.McIntyre RS, Harrison J, Loft H, et al. The effects of vortioxetine on cognitive function in patients with major depressive disorder: a meta-analysis of three randomized controlled trials. Int J Neuropsychopharmacol. 2016;19(10):pyw055. PubMed CrossRef
10.Duman RS, Li N, Liu RJ, et al. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62(1):35-41. PubMed CrossRef
11.Lapidus KAB, Levitch CF, Perez AM, et al. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol Psychiatry. 2014;76(12):970-976. PubMed CrossRef
12.McGirr A, Berlim MT, Bond DJ, et al. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol Med. 2015;45(4):693-704. PubMed CrossRef
13.Quiroz JA, Tamburri P, Deptula D, et al. Efficacy and safety of basimglurant as adjunctive therapy for major depression: a randomized clinical trial. JAMA Psychiatry. 2016;73(7):675-684. PubMed CrossRef
14.Dwyer JM, Lepack AE, Duman RS. mTOR activation is required for the antidepressant effects of mGluR2/3 blockade. Int J Neuropsychopharmacol. 2012;15(4):429-434. PubMed CrossRef
15.Higgins GA, Ballard TM, Kew JN, et al. Pharmacological manipulation of mGlu2 receptors influences cognitive performance in the rodent. Neuropharmacology. 2004;46(7):907-917. PubMed CrossRef
16.Campo B, Kalinichev M, Lambeng N, et al. Characterization of an mGluR2/3 negative allosteric modulator in rodent models of depression. J Neurogenet. 2011;25(4):152-166. PubMed CrossRef
17.Hikichi H, Kaku A, Karasawa J, et al. Stimulation of metabotropic glutamate (mGlu) 2 receptor and blockade of mGlu1 receptor improve social memory impairment elicited by MK-801 in rats. J Pharmacol Sci. 2013;122(1):10-16. PubMed CrossRef
18.Lundström L, Bissantz C, Beck J, et al. Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: mechanistic studies with new potent negative allosteric modulators. Br J Pharmacol. 2011;164(2b):521-537. PubMed CrossRef
19.International Conference on Harmonisation (ICH): Good clinical practice E6 (R1). ICH website. https://www.ich.org/page/efficacy-guidelines. 1996. Accessed January 8, 2018.
20.Chandler GM, Iosifescu DV, Pollack MH, et al. RESEARCH: Validation of the Massachusetts General Hospital Antidepressant History Questionnaire (ATRQ). CNS Neurosci Ther. 2010;16:322-325. PubMed CrossRef
21.Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382-389. PubMed CrossRef
22.Guy W. Clinical Global Impressions. ECDEU Assessment Manual for Psychopharmacology. DHEW Publication No. 76-338. Rockville, MD: National Institute of Mental Health; 1976:217-222.
23.Sahakian BJ, Owen AM. Computerized assessment in neuropsychiatry using CANTAB: discussion paper. J R Soc Med. 1992;85(7):399-402. PubMed
24.Williams JBW, Kobak KA. Development and reliability of a structured interview guide for the Montgomery Asberg Depression Rating Scale (SIGMA). Br J Psychiatry. 2008;192(1):52-58. PubMed CrossRef
25.Trivedi MH, Rush AJ, Ibrahim HM, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med. 2004;34(1):73-82. PubMed CrossRef
26.Fava M, Iosifescu DV, Pedrelli P, et al. Reliability and validity of the Massachusetts General Hospital Cognitive and Physical Functioning Questionnaire. Psychother Psychosom. 2009;78(2):91-97. PubMed CrossRef
27.Sheehan DV. The Anxiety Disease. New York, NY: Scribner; 1983.
28.Endicott J, Nee J, Harrison W, et al. Quality of Life Enjoyment and Satisfaction Questionnaire: a new measure. Psychopharmacol Bull. 1993;29(2):321-326. PubMed
29.Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266-1277. PubMed CrossRef
30.Chouinard G, Margolese HC. Manual for the Extrapyramidal Symptom Rating Scale (ESRS). Schizophr Res. 2005;76(2-3):247-265. PubMed CrossRef
31.Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133(5):429-435. PubMed CrossRef
32.Uher R, Perlis RH, Henigsberg N, et al. Depression symptom dimensions as predictors of antidepressant treatment outcome: replicable evidence for interest-activity symptoms. Psychol Med. 2012;42(5):967-980. PubMed CrossRef
33.Stein DJ, Baldwin DS, Dolberg OT, et al. Which factors predict placebo response in anxiety disorders and major depression? an analysis of placebo-controlled studies of escitalopram. J Clin Psychiatry. 2006;67(11):1741-1746. PubMed CrossRef
34.Chen JA, Papakostas GI, Youn SJ, et al. Association between patient beliefs regarding assigned treatment and clinical response: reanalysis of data from the Hypericum Depression Trial Study Group. J Clin Psychiatry. 2011;72(12):1669-1676. PubMed CrossRef
35.Iovieno N, Papakostas GI. Correlation between different levels of placebo response rate and clinical trial outcome in major depressive disorder: a meta-analysis. J Clin Psychiatry. 2012;73(10):1300-1306. PubMed CrossRef
36.Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. 2013;170(7):723-733. PubMed CrossRef
37.Papakostas GI, טstergaard SD, Iovieno N. The nature of placebo response in clinical studies of major depressive disorder. J Clin Psychiatry. 2015;76(4):456-466. PubMed CrossRef
38.Khan A, Fahl Mar K, Faucett J, et al. Has the rising placebo response impacted antidepressant clinical trial outcome? data from the US Food and Drug Administration 1987-2013. World Psychiatry. 2017;16(2):181-192. PubMed CrossRef
39.Kobak KA, Leuchter A, DeBrota D, et al. Site versus centralized raters in a clinical depression trial: impact on patient selection and placebo response. J Clin Psychopharmacol. 2010;30(2):193-197. PubMed CrossRef
40.Freeman MP, Pooley J, Flynn MJ, et al. Guarding the gate: remote structured assessments to enhance enrollment precision in depression trials. J Clin Psychopharmacol. 2017;37(2):176-181. PubMed CrossRef
41.Khan A, Kolts RL, Thase ME, et al. Research design features and patient characteristics associated with the outcome of antidepressant clinical trials. Am J Psychiatry. 2004;161(11):2045-2049. PubMed CrossRef
42.Fava GA, Guidi J, Rafanelli C, et al. The clinical inadequacy of the placebo model and the development of an alternative conceptual framework. Psychother Psychosom. 2017;86(6):332-340. PubMed CrossRef
43.Ratti E, Bettica P, Alexander R, et al. Full central neurokinin-1 receptor blockade is required for efficacy in depression: evidence from orvepitant clinical studies. J Psychopharmacol. 2013;27(5):424-434. PubMed CrossRef
44.Thase ME, Trivedi MH, Nelson JC, et al. Examining the efficacy of adjunctive aripiprazole in major depressive disorder: a pooled analysis of 2 studies. Prim Care Companion J Clin Psychiatry. 2008;10(6):440-447. PubMed CrossRef
45.Trivedi MH, Thase ME, Osuntokun O, et al. An integrated analysis of olanzapine/fluoxetine combination in clinical trials of treatment-resistant depression. J Clin Psychiatry. 2009;70(3):387-396. PubMed CrossRef
This PDF is free for all visitors!