Objective: To present a review of the literature on the clinical presentation and pathophysiology of anti-N-methyl-d-aspartate receptor encephalopathy (ANMDARE) with attention to both the more commonly recognized psychotic symptom prodrome and the less well-understood depressive symptom prodrome.
Data Sources: The search for clinical neuropsychiatric phenomena and proposed mechanisms involved in ANMDARE pathophysiology was conducted in PubMed. English-language articles published up to September 2019 were identified using a combination of the following search terms: N-methyl-d-aspartate, anti-NMDA receptor encephalitis, schizophrenia, psychosis, depression, major depressive disorder, bipolar I disorder, bipolar II disorder, anxiety, and posttraumatic stress disorder.
Study Selection: From 150 articles identified from the initial search, the 73 most relevant clinical studies, reviews, and case reports related to the study objectives were included.
Data Extraction: Sources were individually analyzed by the 3 authors for the most clinically relevant information.
Results: The pathophysiology and mechanisms involved in anti-NMDA receptor antibody delivery to the brain are incompletely characterized, but antibody binding appears to involve the GluN1 subunit in most cases. Psychotic symptoms are the most commonly recognized components of prodromal psychiatric illness in ANMDARE, which may lead to an initial diagnosis of schizophrenia. In addition to psychotic symptoms, there are reports of depressive symptoms occurring before the emergence of, co-occurring with, or instead of psychotic symptoms in ANMDARE.
Conclusions: In addition to the better-known psychotic prodrome, depressive symptomatology can occur in ANMDARE patients. ANMDARE should be considered in patients with initial presentation of either psychotic or atypical depressive illnesses. Early recognition of these psychiatric prodromal states as antecedents to ANMDARE could lead to improved diagnosis and better management of this potentially life-threatening autoimmune disorder.
This CME activity is expired. For more CME activities, visit CMEInstitute.com.
Find more articles on this and other psychiatry and CNS topics:
The Journal of Clinical Psychiatry
The Primary Care Companion for CNS Disorders

ABSTRACT
Objective: To present a review of the literature on the clinical presentation and pathophysiology of anti-N-methyl-d-aspartate receptor encephalopathy (ANMDARE) with attention to both the more commonly recognized psychotic symptom prodrome and the less well-understood depressive symptom prodrome.
Data Sources: The search for clinical neuropsychiatric phenomena and proposed mechanisms involved in ANMDARE pathophysiology was conducted in PubMed. English-language articles published up to September 2019 were identified using a combination of the following search terms: N-methyl-d-aspartate, anti-NMDA receptor encephalitis, schizophrenia, psychosis, depression, major depressive disorder, bipolar I disorder, bipolar II disorder, anxiety, and posttraumatic stress disorder.
Study Selection: From 150 articles identified from the initial search, the 73 most relevant clinical studies, reviews, and case reports related to the study objectives were included.
Data Extraction: Sources were individually analyzed by the 3 authors for the most clinically relevant information.
Results: The pathophysiology and mechanisms involved in anti-NMDA receptor antibody delivery to the brain are incompletely characterized, but antibody binding appears to involve the GluN1 subunit in most cases. Psychotic symptoms are the most commonly recognized components of prodromal psychiatric illness in ANMDARE, which may lead to an initial diagnosis of schizophrenia. In addition to psychotic symptoms, there are reports of depressive symptoms occurring before the emergence of, co-occurring with, or instead of psychotic symptoms in ANMDARE.
Conclusions: In addition to the better-known psychotic prodrome, depressive symptomatology can occur in ANMDARE patients. ANMDARE should be considered in patients with initial presentation of either psychotic or atypical depressive illnesses. Early recognition of these psychiatric prodromal states as antecedents to ANMDARE could lead to improved diagnosis and better management of this potentially life-threatening autoimmune disorder.
Prim Care Companion CNS Disord 2020;22(3):19r02563
To cite: Nguyen AV, Young KA, Bourgeois JA. Psychiatric prodrome in anti-NMDAR-associated encephalopathy: clinical and pathophysiologic considerations. Prim Care Companion CNS Disord. 2020;22(3):19r02563.
To share: https://doi.org/10.4088/PCC.19r02563
© Copyright 2020 Physicians Postgraduate Press, Inc.
aDepartment of Psychiatry, Baylor Scott & White Health, Central Texas Division and College of Medicine, Texas A&M University Health Science Center, Temple, Texas
*Corresponding author: James A. Bourgeois, OD, MD, FACLP, Department of Psychiatry, Baylor Scott & White Health, Central Texas Division, 2401 South 31st St, Temple TX 76508 ([email protected]).
Glutamate neurons form the backbone for long-distance neurotransmission in the brain, making glutamatergic synaptic transmission vital for learning, memory, emotion, and behavior.1,2 Disturbances in the glutamatergic system are thought to be critical components of a variety of neuropsychiatric syndromes. The N-methyl-d-aspartate receptor (NMDAR) is an important pre- and postsynaptic component of this system.3 The brain is an immune-privileged organ, but in rare situations, autoantibodies to the NMDAR may develop and produce anti-NMDAR encephalitis (ANMDARE) by attacking neurons. Autoantibodies targeting the NMDAR may produce psychotic or depressive symptoms before more severe, life-threatening neurologic symptoms appear. These symptoms may thus represent a “psychiatric prodrome.” Over two-thirds of ANMDARE patients experience psychiatric symptoms and initially present for psychiatric care.4 In light of the emerging literature on the psychiatric presentation of ANMDARE, the authors reviewed the recent pertinent literature to provide clinicians with guidance on identification and management of possible ANMDARE cases with a primarily psychiatric presentation as a psychotic or depressive disorder.
METHODS
The search for clinical neuropsychiatric phenomena (primarily psychotic and depressive disorders) and proposed molecular and receptor mechanisms involved in ANMDARE pathophysiology was performed using PubMed for English-language articles up to September 2019. The search terms used were combinations of the following: N-methyl-d-aspartate, anti-NMDA receptor encephalitis, schizophrenia, psychosis, depression, major depressive disorder, bipolar I disorder, bipolar II disorder, anxiety, and posttraumatic stress disorder. From the 150 articles identified from the initial search, all abstracts were reviewed by the authors for greatest relevance to the clinical presentation of ANMDARE with a psychotic or depressive disorders presentation. After this initial review, the 73 most relevant remaining clinical studies, reviews, and case reports1-73 related to the study objectives were included.
Typical ANMDARE Clinical Syndrome
Autoimmune causes of psychiatric symptoms date back to 1937 when Lehmann-Facius proposed that schizophrenia was caused by autoantibodies targeting brain tissue.5 The first proposed case of autoimmune encephalitis was identified in 1968 as limbic encephalitis associated with small-cell lung cancer.6 A form of limbic encephalitis with immunoglobulin G (IgG) antibodies against voltage-gated potassium channels was subsequently characterized in 2001.7 In 2005, a case report8 described 7 patients with subacute limbic encephalitis of uncertain etiology, 6 of whom demonstrated remarkable clinical improvement after immunotherapy or tumor resection. An ovarian teratoma was found in one of those patients.8 Dalmau et al9 proceeded to characterize the first cases of ANMDARE in 2007 when they described a severe but treatment-responsive encephalitis in 12 women. Since then, over 1,000 ANMDARE cases have been reported. Although ANMDARE is the most common autoimmune encephalitis disorder discovered to date, autoantibodies have been reported for at least 15 other proteins and receptors, including the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) and GABAB (γ-aminobutyric acid) receptors.3,9,10
ANMDARE symptoms typically progress through distinct phases. The disease usually starts with a prodromal phase characterized by constitutional, “flu-like” symptoms often with unrecognized subtle psychiatric symptoms such as anxiety, agitation, short-term memory loss, and isolated performance deficits.11 This phase is followed by 1-3 weeks of more severe and debilitating psychiatric symptoms, with the third phase leading to the development of neurologic symptoms including dyskinesia, seizures, and autonomic instability.11-13 However, some cases have initially presented with severe neurologic symptomatology needing intensive care unit admission and intubation, with no obvious preceding psychiatric disturbance.14 A study by Herken and Pr×¼ss15 attempting to identify “red flag” symptoms of ANMDARE found that 23 of 53 (43%) patients experienced hallucinations, while seizures were present in 10 of 53 (19%)patients. Decreased levels of consciousness or paralysis were present in 9 of 53 (17%) patients (Table 1).
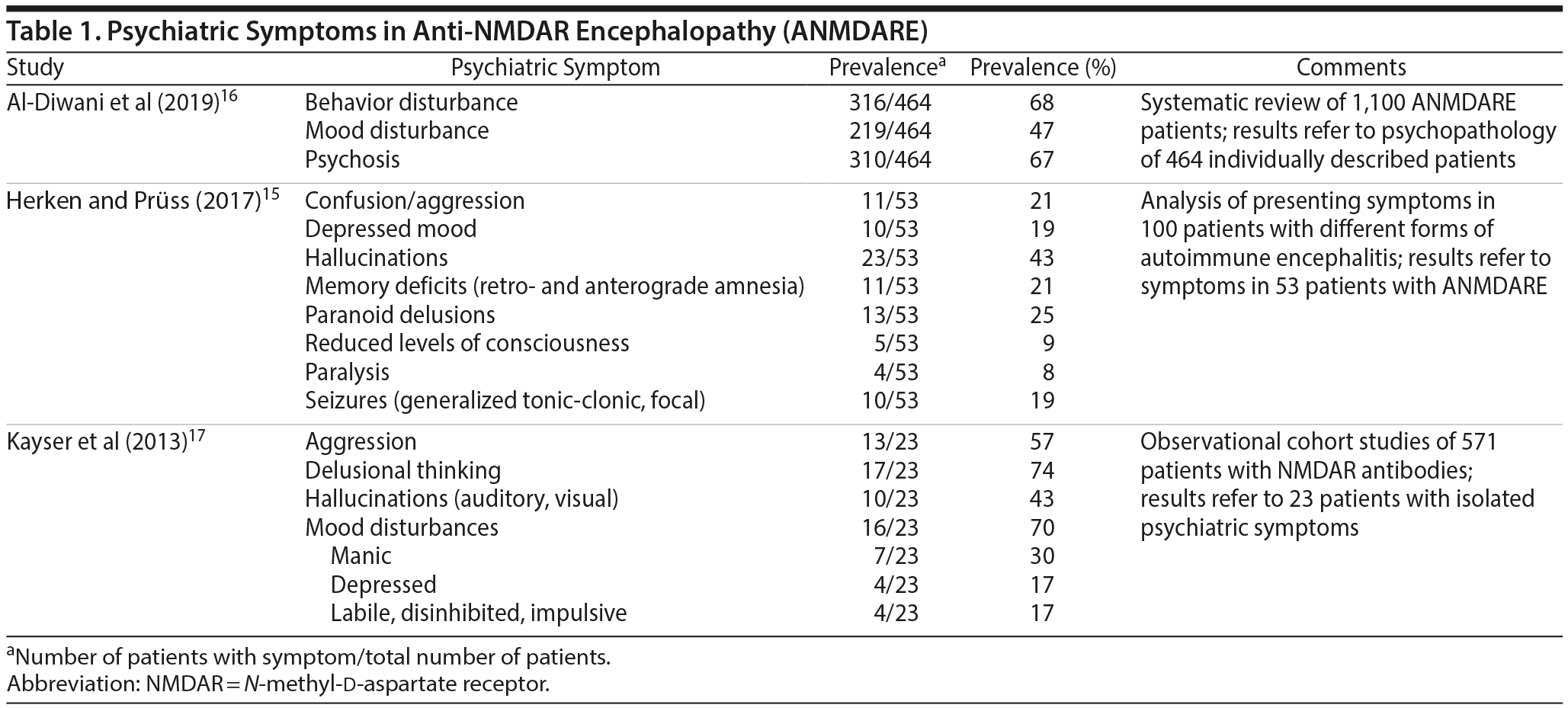
A systematic review by Al-Diwani et al16 of 464 ANMDARE patients found that 67% displayed psychotic symptoms (Table 1). Delusions were the most common psychotic symptoms, followed by auditory and visual hallucinations. Kayser et al17 reported that early psychiatric presentations in ANMDARE included delusions (74%), auditory or visual hallucinations (43%), and aggressive behavior (57%) (Table 1). ANMDARE is thus frequently initially diagnosed as a psychotic disorder, eg, schizophrenia, drug-induced psychotic disorder, or postpartum psychotic disorder.12,18-20 While the eventual development of neurologic symptoms is the most common longitudinal course for ANMDARE, some patients never progress beyond a purely psychiatric presentation. In the study of 571 ANMDARE patients by Kayser et al,17 23 had a course of illness that did not include neurologic symptoms. Five of the 23 patients had persistent psychiatric symptoms without recovery, while the remaining 18 had a remitting/relapsing psychiatric course. This finding supports the possibility that ANMDARE could be an unrecognized cause of psychotic illness.17
While a direct relationship between anti-NMDAR antibodies and psychiatric symptoms may exist, more complex mechanisms may be needed to explain observations of anti-NMDAR autoantibodies in normal controls. A large study of 4,236 individuals by Dahm et al21 found comparable levels of NMDAR1 autoantibodies among healthy (8.5%, 145/1,703) and neuropsychiatrically ill patients (11%, 278/2,533; the study population included schizophrenia, affective disorders, stroke, Parkinson disease, amyotrophic lateral sclerosis, and personality disorders). Similarly, in a large study of 2,817 subjects (1,325 healthy controls; 1,081 schizophrenia, 263 Parkinson disease, and 148 affective disorder patients including those with major depressive disorder and bipolar disorder), Hammer et al22 found that 10.5% of participating patients were seropositive for antibodies against the NR1 subunit of NMDAR. Surprisingly, 143 healthy controls (12%) were also seropositive for anti-NMDAR antibodies.22
The prevalence of NMDA antibodies in normal controls is unexplained. Hammer et al22 proposed that various factors, such as the presence of a compromised blood-brain barrier, may predispose certain individuals to passage of blood-borne antibodies into the brain, leading to central nervous system (CNS) effects. The finding that injection of anti-NMDAR antibodies in ApoE mice with known blood-brain barrier leakage, but not in control mice, caused psychosis-like behavioral changes supports this theory.22 A “two hit” hypothesis for NMDA pathology may need to be developed to explain these animal data and the nontrivial reported incidence of NMDAR antibodies found in the blood of healthy subjects. Neuropathology would require not only the development of NMDA antibody production by peripheral B-cells but also additional conditions such as a blood-brain barrier deficit to alter the penetration of blood-borne antibodies into the brain or changes in B-cell penetration and clonal expansion in the intrathecal space.
Diagnostic Testing and Outcomes
The diagnosis of ANMDARE is confirmed by detection of IgG antibodies against the GluN1 subunit of the NMDAR. Cerebrospinal fluid (CSF) IgG analysis is more sensitive and specific when compared to serum; consequently, it would be rare to have positive serum with negative CSF findings.23 Although tests are highly sensitive and specific for ANMDARE, IgG testing can take days to weeks for results. Early detection and treatment of the disease is necessary to prevent long-term sequelae. A systematic review by Blinder and Lewerenz23 characterized basic CSF findings in various subtypes of autoimmune encephalitis (AIE), including ANMDARE. ANMDARE CSF is usually inflammatory and characterized by increased protein, pleocytosis, and the presence of CSF-restricted oligoclonal bands. Pathological protein levels > 1,000 mg/L were detected in 4 of 18 (22%) tested ANMDARE patients. A relative pleocytosis of ≥ 20 cells/µL was found in > 60% of ANMDARE patients, with a pleocytosis of > 100 cells/µL found in 18 of 52 (35%) ANMDARE patients. This pattern was found to be more specific to ANMDARE, as inflammatory CSF changes are rarer in other AIE subtypes (eg, AIE associated with LGI1 antibodies).23
In multivariable analyses performed by Gresa-Arribas et al,24 CSF and serum titers were higher in patients with poor outcomes than in those with good outcomes (CSF dilution 340 vs 129, P = .049; serum 7,370 vs 1,243, P = .0025). Clinical outcome was determined using the modified Rankin Scale (mRS). Good outcome (mRS = 0-2) and poor outcome (mRS > 2) were determined at the patient’s last follow-up (median of 26 months). Of note is that decreases in antibody titers were observed across all patients regardless of outcome.24
Positive testing for NMDAR antibodies does not always correlate with disease and can obscure treatment of other diseases. Serum and CSF that were strongly positive for NMDAR antibodies has been reported in a patient with Lyme neuroborreliosis, a CNS infection caused by Borrelia burgdorferi.25 This patient, despite having positive titers, had a clinical picture incompatible with ANMDARE and demonstrated no signs of impaired cognition during hospital admission.25
Pathophysiology
There is substantial pharmacologic, genetic, and biochemical evidence supporting the hypothesis that NMDAR hypofunction mediates psychotic symptoms.26 NMDAR antagonists such as phencyclidine and ketamine have been shown to induce psychotic symptoms as well as behavioral and cognitive impairments, similar to symptoms observed in schizophrenia and early ANMDARE. NMDAR hypofunction on inhibitory interneurons is thought to be an initial step in ANMDARE, with loss of inhibitory tone on glutamate neurons leading to corticolimbic disinhibition.1,27 Excessive glutamate release in prefrontal and subcortical structures could thus contribute to the psychosis and bizarre dyskinesias observed in ANMDARE.28 The localization of ANMDAR staining in the cortex and hippocampus is consistent with observed patterns of patient symptomatology.13,29
The NMDAR is a glutamate receptor composed of 4 subunits that typically includes 2 GluN1 subunits in combination with either 2 GluN2 or 1 GluN2 and 1 GluN3 subunit.30 These components form a ligand-gated cation complex that is highly permeable to calcium. Previous studies30 have demonstrated that there are 8 splice variants of GluN1 (GluN1-1a to GluN1-4a and GluN1-1b to GluN1-4b), 4 variants of GluN2 subunits (2A-2D), and 2 variants of the GluN3 subunits (3A-B).
- Atypical and treatment-resistant depressive symptomatology can occur in anti-N-methyl-d-aspartate receptor encephalopathy (ANMDARE), and early recognition could improve disease management and patient outcomes.
- NMDAR antibodies have been found in healthy and neuropsychiatrically ill patients; a clinical correlation must be made, and a positive result does not necessarily correlate with encephalitis.
- Treatment of ANMDARE should be individualized and emphasize multispecialty care.
Dalmau et al13 proposed that NMDAR antibody binding was selective for the GluN1 subunit of the NMDAR. In this study,13 molecular alterations to the GluN1 subunit, and not the GluN2 subunit, were found to be associated with changes in the level of NMDA antibody binding. Subsequent research using a plasmid (NR1d4) encoding a GluN1subunit without amino acid residues 25 to 380, but still able to bind GluN2, showed decreased reactivity to NMDAR antibodies. This finding led to the discovery of an extracellular region of the GluN1 subunit that was universally recognized by patient-derived NMDAR antibodies.2,13 Further in-depth studies29 have shown that the N368/G369 residues of GluN1 are critical for antibody recognition and creation of immunoreactivity. This amino terminal domain (ATD) of GluN1 is required for antibody binding.29,31
GluN subunits are composed of the ATD, S1, and S2 domains, which form a ligand-binding domain, 3 membrane-spanning domains (TM1, 3, 4), a membrane loop (TM2), and an intracellular C-terminal domain that links to scaffolding proteins and messenger systems.29 The ATD of GluN1 has 7 N-linked glycosylation sites. Patient antibodies do not stain cells when glycosylation is blocked by tunicamycin, an N-acetylglucosamine transferase inhibitor, suggesting that glycosylation is required for epitope creation. However, it is important to note that tunicamycin impacts NMDAR expression and formation.32 Further studies29 were performed by mutating each of the 7 glycosylation sites to make them impervious to glycosylation; of the 7 mutants, only the N368 mutant affected antibody staining. This finding suggests that glycosylation most likely contributes to pathogenesis by creating favorable conformations of the N368 region of the GluN1 ATD, allowing NMDA antibodies to bind.
The leading theory of anti-NMDAR antibody pathophysiology in the brain holds that NMDAR antibody binding induces loss of synaptic NDMARs through receptor internalization. This theory is supported by multiple lines of evidence. Anti-NMDAR antibodies bind, cap, and cross-link NMDARs, resulting in receptor internalization and reductions in synaptic NMDAR surface density, and human anti-NMDAR antibodies deposited in the rat hippocampus selectively and reversibly decrease the surface density of synaptic NMDARs.33 This process occurs without loss of other synaptic components or neuron viability and is dose dependent. This finding is supported by in vivo and in vitro immunohistochemical observations, as well as in vivo electrophysiology findings. For instance, decreases in NMDAR-mediated synaptic currents have been observed in antibody-treated rat hippocampi.2,33 Subsequent studies, also in rats, found that the decrease in NMDAR surface density plateaus 12 hours after antibody administration and persists for as long as there is antibody present.1 These findings provide strong support indicating that reduced NMDA receptor density may be a primary cellular mechanism responsible for ANMDARE. Receptor endocytosis may subsequently lead to functional inhibition of NMDARs, which would explain the prominent psychosis seen in ANMDARE that is similar to the effects of NMDA antagonists such as phencyclidine and ketamine.
An important aspect of inhibitory tone in the cortex and hippocampus involves the activation of interneurons through excitation of postsynaptic glutamate receptors. In some interneuron subtypes, these glutamate receptors are largely composed of NMDARs.34 Loss of NMDA receptors on GABA neurons in ANMDARE may prevent these inhibitory cells from being activated by glutamate, which produces an overall reduction in inhibitory tone. Loss of inhibitory constraints on glutamate neurons can result in hyperactivity mediated by postsynaptic signaling through other glutamate receptor subtypes not affected by anti-NMDA antibodies, such as AMPA receptors.35 Consistent with this scenario, reductions in extracellular levels of GABA and elevations in extracellular glutamate have been observed in ANMDARE.36 Loss of inhibitory tone and glutamate hyperactivation may explain symptoms such as seizures. This glutamate-induced excitotoxicity occurs at high antibody concentrations in a dose-dependent manner through enhancement of mitochondrial permeability.37 Since differential dose effects on these inhibitory/excitatory interactions have been reported, low and high doses of antibodies may contribute to different ANMDARE neuropsychiatric symptoms and syndromes.37,38
Although systematic studies are lacking, it is expected that ANMDAR antibodies have selected effects that may include inflammatory processes in different areas of the brain. A postmortem microscopic study by Tuzun et al39 suggests a prominent role of antibody immune response and inflammation in ANMDARE pathophysiology. This study39 found increased reactive microglia in all areas of the CNS in 2 ANMDARE patients. Microgliosis was highest in the hippocampus, forebrain, basal ganglia, and spinal cord. It was also observed that cytotoxic T-cell mechanisms were not as prominent as detection of cells expressing cytotoxicity markers (TIA, granzyme B, perforin, Fas/Fas ligand).39 Inflammation of the hippocampus and medial temporal lobe was observed during autopsy of a young woman with refractory ANMDARE who had a history of herpes simplex virus (HSV) infection.40 Notably, HSV infection was thought to not have caused the hippocampal or temporal lobe inflammation, since this patient had numerous negative infection assays, including CSF HSV analysis by polymerase chain reaction and enzyme-linked immunosorbent assay.40 Atrophy of the temporal lobes and hippocampi were also observed with magnetic resonance imaging (MRI) and postmortem examination of a 35-year-old woman with suspected ovarian teratoma-related ANMDARE.9 Postmortem analysis identified significant loss of hippocampal pyramidal neurons with extensive gliosis and microglial proliferation in this case. Immunohistochemistry subsequently demonstrated moderate lymphocytic infiltrates in the parahippocampus, thalamus, insula, and medulla.9 The possible role of immune and inflammatory processes and brain localization of pathology in ANMDARE are topics that need to be further explored.
Few studies have attempted to correlate ANMDARE-related symptoms with regional deficits or brain oscillatory activity. Network dysfunction in ANMDARE is evident with electroencephalogram changes such as extreme delta brush. Other imaging modalities, such as 18F-fluorodeoxyglucose positron emission tomography, have identified diffuse cortical hypometabolism with specific involvement of the temporal lobe in cases with relapsing ANMDARE.41,42 Alterations in the hippocampal and temporal lobes could contribute to psychotic symptoms, as these brain regions have been implicated in schizophrenia and bipolar disorder.43 In addition to these areas, anti-NMDA antibodies could affect the amygdala, an important structure in the limbic system intimately linked with emotional processing. Melzer et al44 proposed that autoantibody action in the basolateral complex of the amygdala, the main input site for sensory information from the thalamus and cortex, could contribute to neuropsychiatric symptoms in ANMDARE. Only 55% of 100 patients with encephalitis and NMDAR antibodies displayed increased MRI fluid-attenuated inversion recovery or T2 signal in ≥ 1 brain regions, and these findings did not significantly correlate with symptoms.13 Spike discharge patterns are important for neuronal coding of information, and these signals include high-frequency patterns such as γ oscillations with frequencies between 30 and 90 Hz.45 Although there are no studies of γ oscillations in ANMDARE patients, NMDA antagonists such as ketamine and MK801 dose-dependently increase wake-related γ oscillations.36 It is proposed that the increase in γ oscillations produced by NMDA antagonists may generate symptoms by disrupting conscious integration.36 Further research into regional changes in γ oscillations and their influence on functional connectivity may prove useful in diagnosing and monitoring treatment in ANMDARE.
Early researchers recognized that ANMDARE was associated in many cases with ovarian teratomas, which can express many neuronal genes including NMDARs.3,9 As the cancer tissue necrotizes, it enters the lymphatic system and is delivered to the lymph nodes wherein immune cells can generate receptor antibodies. It is thought that ectopic expression of the NMDAR and subsequent delivery of large amounts of receptor to the attention of the immune system contributes to receptor sensitization and loss of immune tolerance to the NMDAR in teratoma-associated ANMDARE. NMDAR proteins generated in teratomas may by abnormally translated and differ from those generated in the CNS, and these proteins could thus be sensed as “foreign” by the immune system. Neural tissue derived from ovarian teratomas in healthy patients without anti-NMDAR antibodies have also been observed to react with IgG from patients with NMDAR antibodies.46 Plasma cells with NMDA antibodies generated by the immune system may then be released into the blood where they traffic to the intrathecal space, undergoing clonal expansion with resulting release of antibodies into the CSF.47,48 Autopsies have identified NMDAR-positive plasma cells in the intrathecal space, making this form of CNS delivery of antibodies a possible contributor to ANMDARE.47 CSF antibody titers are usually higher than blood levels, potentially facilitating the diffusion of antibodies into the parenchyma. In addition to a presence in the CSF, plasma cells have been identified in the brain lymphatic system and in the white or gray matter of ANMDARE patients.40,49,50
Symptom patterns in ANMDARE suggest 2 distinct phases of the illness. There are often 10-20 days when early psychiatric symptoms antedate movement disorders and dysautonomia. Irani et al48 proposed that this early phase is related to initial intrathecal penetration and antibody diffusion into cortical and hippocampal gray matter. At low doses, antibodies may target interneurons with resulting disinhibition and psychiatric symptoms. The late phase may emerge as a secondary immunologic expansion within the intrathecal compartment, which could generate higher doses of antibodies that further disturb glutamatergic neurotransmission and increase neurologic symptoms.48
Despite mounting evidence suggesting plasma cell presence in the CNS in many ANMDARE cases, there is evidence that B-cells or plasma cells may not need to be present in the CSF or brain for antibodies to penetrate into the brain. Hippocampal and temporal inflammation with T-cell and microglial involvement was observed in a patient who died after 3 months of rituximab immunotherapy even though few plasma cells or plasmablasts could be identified in the spinal cord or CNS upon autopsy.40 Low levels of circulating B-cells before death documented effective B-cell therapy. Despite the lack of circulating or brain B-cells, this patient had a high titer of ANMDAR antibodies in the plasma just before death, and abundant plasma cells were identified in the lymph nodes postmortem. This case suggests that antibody-producing plasma cells can remain in lymphoid tissues for extended periods of time even after immunotherapy and that peripheral sources of antibodies may contribute to encephalopathy.40
In further support of a role for blood-borne antibodies in ANMDARE, levels of blood anti-NMDAR antibodies fluctuate over time, and titers are strongly correlated with symptoms. In a study of 60 ANMDARE patients with manic episodes, Dickerson et al51 found that patients had elevated blood levels of anti-NMDA antibodies at admission (t = 2.99, P = .003) and at clinical evaluation (t = 2.57, P = .01) but not after remission 6 months later. A similar pattern was observed in a 28-year-old woman with ANMDARE who presented with delusions and grandiosity. Elevated anti-NMDA antibody titers were present in this case on relapse (1:64) when compared to initial presentation (1:8).17,52 Further research is needed to determine whether these antibodies present in blood have escaped from the CSF or whether they are generated in the periphery and penetrate into the brain to contribute to ANMDARE pathophysiology.
Autoimmune Comorbidities
ANMDARE has been linked to ongoing HSV infections. Up to 20% of HSV encephalitis patients develop antibodies against various cell surface proteins including the NMDAR.3 Unlike the primarily psychiatric initial presentations in adults, herpes-associated ANMDARE in children initially manifests primarily as choreoathetosis.53,54 Defining features of herpes-associated ANMDARE include focal electroencephalogram abnormalities and positive HSV CSF polymerase chain reaction.55 Although there are no studies that definitively demonstrate the mechanism by which HSV induces NMDAR antibody formation, proof-of-concept studies performed by Linnoila et al56 have shown that denovo NMDA antibody synthesis occurs in mice inoculated with HSV. This process led to decreased hippocampal postsynaptic NMDAR clusters and hippocampal NMDAR protein, similar to that observed in ANMDARE,56 suggesting that HSV- and teratoma-associated ANMDARE may produce similar brain pathophysiology. A study by Kothur et al57 identified a robust inflammatory syndrome in post-herpes simplex encephalitis ANMDARE associated with increased T-helper cells, B-cell-mediated cytokines (CXCL13, CCL19, APRIL), and interferon α. CXCL13, CCL19, and APRIL may contribute to NMDA antibody formation since they facilitate B-cell recruitment, proliferation, and clonal expansion.57 Future studies of the relationship between HSV infection and ANMDARE will be necessary to more fully establish the biological mechanisms responsible for this and other ANMDARE comorbidities.
Depressive Symptoms in ANMDARE
In addition to the occurrence of prominent psychotic symptoms, widely recognized as prodromal before the emergence of neurologic symptoms in ANMDARE, depressive symptoms may also occur in this syndrome. There is evidence that depressed mood and dysphoric affect are often present in the early stages of ANMDARE. Identification of depressive symptoms as an early sign of ANMDARE represents a missed opportunity to improve early diagnosis of this syndrome. Thus, this is particularly important for clinicians to consider, since there are cases of depressive symptoms alone occurring early in ANMDARE before progression to the motor/neurologic phase of the syndrome.
In a study of 86 patients with positive NMDAR antibodies, 22 of whom met diagnostic criteria for ANMDARE, Gibson et al58 found that patients with psychotic features had a characteristic presentation consisting of severe and disproportionate cognitive disturbance (P < .005) with high negative symptom load and behavioral hyperexcitability. The Positive and Negative Symptom Scale measures increasing symptom severity on a 1 to 7 scale, and ANMDARE patients demonstrated a predominance of negative compared to positive symptoms, with mean scores of approximately 5 and 3, respectively.58 Positive symptoms indicate “additional” or “intrusive” symptoms such as hallucinations or delusions, while negative symptoms imply a deficit or absence of a behavior, such as loss of interest (apathy) or loss of a sense of pleasure (anhedonia). Negative symptoms in this instance included blunted affect, emotional withdrawal, poor interpersonal rapport, lack of spontaneity, passive social withdrawal, and motor retardation. Further analysis with the Scale for the Assessment of Negative Symptoms (0 to 5 scale) showed prominent alogia and attention deficits, with mean scores of approximately 4 and 4.5, respectively.58 In another study, Al-Diwani et al16 reported that 219 of 464 (47%) ANMDARE patients displayed depressed and unstable mood (Table 1). These data highlight the presence of prominent negative symptoms, usually associated with depressive syndromes, in the ANMDARE prodromal phase.
Case reports and series also support the possibility that depressive disorder may be a common but unrecognized early harbinger of ANMDARE. Herken and Pr×¼ss15 reported that depressed mood occurred as an initial symptom in 10 of 53 (19%) studied ANMDARE patients (Table 1). For example, emergence of depressive symptoms preceded psychotic symptoms by 2 months in the case of a 16-year-old girl who initially presented with fatigue, sadness, and hypersomnia.59 This case eventually progressed to psychotic symptomatology with auditory and visual hallucinations, resulting in a differential diagnosis of psychotic disorder/major depressive disorder with psychotic features. Eventually, onset of neurologic symptoms occurred and ANMDARE was identified.59 Mixed depressive and manic symptoms with prominent psychotic features are a hallmark of severe bipolar disorder, and ANMDARE patients are sometimes diagnosed with this psychiatric disorder. Mantere et al60 reported that 6/70 (8.6%) ANMDARE patients originally were diagnosed with bipolar disorder. A 32-year-old woman presented with new-onset depressive disorder 10 years after initial symptoms and 6 years after treatment for ANMDARE.61 In a 15-year-old girl who initially presented with restlessness and hallucinations, depressive symptoms appeared a year after treatment.14 Finally, it is worth noting that even when the primary ANMDARE psychiatric presentation is a depressive disorder, psychotic features may be comorbid, as was the case of a 23-year-old man with recurrent major depressive episodes leading to psychiatric treatment exclusively, before an eventual diagnosis of ANMDARE.60 In this case, paranoid fears and illusions were also reported.60
While depressive symptoms appear to precede or accompany psychotic symptoms in many cases of ANMDARE, there are only a few instances of ANMDARE diagnosis based on depressive symptoms alone, possibly because depressive disorders are very common in the general population. Researchers62 have pointed out that since there are many cases of ANMDARE in which psychotic presentations never progress to neurologic symptoms, it is likely that there are undetected cases of ANMDARE masquerading as bipolar disorder or major depressive disorder.
Depression in ANMDARE is different when compared to its classical presentation as outlined in the DSM-5. ANMDARE typically has a variable neuropsychiatric presentation, and relatively few cases have isolated psychiatric symptoms. As a result, depressive disorders in ANMDARE often present alongside variable neurologic symptoms and frequently require multiple medications to control.63 ANMDARE should thus be in the differential diagnoses for, and may well be overrepresented in, patients with complex and treatment-resistant depressive disorders. Although the exact duration of depressive symptoms in ANMDARE is difficult to determine, many patients experience variable levels of subjective symptoms (eg, memory impairment, attention deficits, executive dysfunction) over the long term following immunomodulation therapy or teratoma resection when applicable.63
Animal models support the involvement of mood changes as part of the early psychiatric syndrome in ANMDARE. In a study by Gao et al,64 anti-NMDAR antibodies in lupus-prone mice were directly correlated with immobility in the swim test, a model of depressive symptomatology. In another study,52 C57BL6/J mice infused with antibodies originating from the serum or CSF of ANMDARE patients developed progressive memory deficits, anhedonia, and depressive-like behaviors, with no other behavioral symptoms. Postmortem analysis of these mice subsequently showed progressive increases in bound human antibodies predominantly in the hippocampus.52 Similar results were produced by W×¼rdemann et al,65 who injected CSF fluid from ANMDARE patients into the dentate gyrus of adult mice. The injections decreased long-term potentiation in the dentate gyrus, which led to severe impairment of learning and depression-like performance in the Morris water maze task. Animal data thus support the involvement of symptoms of depression in the psychiatric prodromes of early ANMDARE.
Symptoms of depressive disorder observed in several autoimmune conditions have been linked to anti-NMDA antibodies. Omdal et al66 observed that anti-NMDA antibody levels were associated with poor performance on several cognitive and psychological tests in systemic lupus erythematosus patients and concluded that anti-NMDAR antibodies might be responsible not only for short-term memory and learning deficits in these patients but also for depressed mood as well. Lapteva et al67 reported that anti-NMDA antibodies were associated with depressed mood in 60 patients with systemic lupus erythematosus. In this study, patients with Beck Depression Inventory scores ≥ 14 had higher serum anti-NMDA antibodies than patients with lower scores. Patients who met DSM-IV criteria for major depressive disorder had a trend toward higher levels of anti-NMDA antibodies.67 In a study68 of 66 patients with primary Sjogren’s syndrome, a higher proportion of depressed patients had anti-NMDA antibodies compared to those without depressive disorders.68 It is interesting that while the GluN1 subunit is most commonly associated with idiopathic or teratoma-associated ANMDARE, depressive symptoms in systemic lupus erythematosus and Sjogren’s syndrome appear to be related to GluN2 antibodies.37,69
In conclusion, there is considerable evidence from both animal models and clinical observations to support the possibility that negative symptoms represent a substantial but largely unrecognized component of the overall symptom load in ANMDARE. Attention to negative symptomatology as a precursor to ANMDARE may provide additional clinical clues to help in the diagnosis of this treatable condition.
Clinical Interventions
Current treatment of ANMDARE involves immunotherapy and careful survey and excision of a neoplasm (if present). Intravenous immunoglobulin (IVIG) and plasma exchange can be utilized to lower autoantibody titers, while corticosteroids can be employed to decrease inflammation. While first-line treatment is usually effective, second-line immunotherapy is often required. In the case of nonresponse or exacerbation of encephalitis, it is also important to remember that high-dose corticosteroids can themselves cause psychiatric symptoms, such as psychosis. Rituximab, which acts against CD20 monoclonal antibodies and suppresses B-cell functions, is often used in refractory ANMDARE cases. In a recent case study of a patient refractory to rituximab treatment, Sveinsson et al70 supplemented rituximab with bortezomib to target plasma cells as well as B-cells and achieved a substantial level of remission with slowly recovering cognitive function and partial reversal of cerebral atrophy. Although the course of ANMDARE varies according to many factors (eg, presence of teratoma, need for intensive care unit management, time from symptom onset), it generally has a relatively positive prognosis. A study2 that followed the treatment of 577 ANMDARE patients found substantial neurologic improvement in 81% of patients after a median follow-up of 24 months.
Warren et al63 explored the psychiatric management in a cohort analysis of 30 adult ANMDARE cases in Queensland, Australia from 2011 to 2018. Most of these patients were treated with a mean olanzapine equivalent dose of 11.5 mg/day prior to initiating immunomodulation. Antipsychotic treatment was associated with an 88% reduction in aggression (7/8 cases), although there was little improvement in psychosis, affective symptoms, or catatonia.63 Of importance is that ANMDARE-associated psychiatric symptoms can respond partially to “routine” psychotropic medications, so improvement with medications does not a priori rule out ANMDARE. Warren et al63 proposed that antipsychotics were not fully effective due to the rapid network dysfunction that occurs in ANMDARE, as well as possible disruption to the mesolimbic dopaminergic pathways. Cognitive recovery in this cohort was found to be variable, as ANMDARE may cause dysfunction in cellular mechanisms associated with learning.63 This dysfunction is compounded by fatigue, poor attention, and slow processing speeds, which may decrease participation and engagement in activities required for recovery. Following immunomodulation and teratoma resection (where applicable), there was complete resolution in 8 cases (27%), partial improvement in 18 cases (60%), and little/no behavioral improvement in 4 cases (13%). Twelve cases required continued antipsychotics, with 8 of these patients requiring long-term treatment to the study’s end point. While encephalitis may recur (2/30 in this cohort), particularly following treatment discontinuation, symptoms usually resolve with reinitiation of therapy.63
Antipsychotic and other psychotropic medications are relatively ineffective in treating psychiatric symptoms in ANMDARE patients.14,20,61,71 Clinicians should thus consider ANMDARE in the differential diagnosis of treatment-resistant psychotic disorders. Resistance has been reported for a wide range of psychotropic medications including lorazepam, lithium, haloperidol, risperidone, and olanzapine.14,71 A 28-year-old woman with psychotic features later identified as ANMDARE related was initially treated with haloperidol. After dosage escalation to 5 mg/day, she developed severe extrapyramidal symptoms.12 Five milligrams is not a high daily dose of haloperidol, and it has been suggested that ANMDARE may increase the risk of developing antipsychotic medication side effects.12 In support of this possibility, the dopamine blockade action of antipsychotics has been demonstrated to exacerbate dyskinetic and dystonic movements when used in agitated ANMDARE patients.11,72,73 However, it is possible that side effect exacerbations could potentially result from oversedation in the setting of an antipsychotic-naive patient.11 Better recognition of ANMDARE as a possible cause of psychotic symptoms could prevent futile exposure to nonproductive and potentially problematic drugs.
Variable and complex psychiatric presentations in ANMDARE necessitate continued psychiatric input. The development of standard protocols for intervention has been limited by small case numbers and a lack of clinical trials. Management should thus be flexible and vary depending on factors such as the type of psychiatric symptom, stage and severity of illness, neurologic symptoms, and medication side effects. It should be emphasized that treatment of ANMDARE must be individualized and involve multispecialty medical care to provide patients with the best possible outcomes.63
CONCLUSIONS
ANMDARE is a potentially lethal autoimmune disorder that is usually associated with early psychiatric symptoms and a later emerging neurologic syndrome. While the triggers for developing autoimmunity are not yet determined, it is clear that IgG antibodies attack NMDARs, particularly the GluN1 subunit in several regions of the brain in ANMDARE. The omnipresence of the GluN1 receptor throughout the brain and its distribution on both glutamate neurons and interneurons may be responsible for the breadth of early psychiatric symptoms in this syndrome. Early psychiatric symptoms may be related to impairment of inhibitory interneurons with resulting disinhibition of principle glutamate projection neurons. As the load of ANMDAR antibodies builds in the brain, grossly impaired neurotransmission in the primary glutamatergic projection neurons of the brain may then lead to late-stage neurologic impairments. Since there are cases wherein antibody-releasing plasma cells are only found in immune organs and not in the brain, and since there is a relatively high incidence of ANMDAR antibody findings in the blood of normal, asymptomatic individuals, a two-hit hypothesis may be needed to explain ANMDARE brain pathophysiology. Early recognition of ANMDARE is important for treatment because intervention can abrogate progression to late-stage neurologic complications. CSF IgG analysis is preferred over serum for the diagnosis of ANMDARE, but a positive result does not always correlate with encephalitis.
Treatment should be individualized and emphasize a multidisciplinary approach. Isolated psychiatric presentations are rare, and ANMDARE may be overrepresented in complex and treatment-resistant depression. Although ANMDARE-associated psychiatric symptoms are less responsive to routine psychotropic medications, a partial response may be observed. Most documented cases of ANMDARE are associated with a prodrome that includes prominent psychotic symptoms, and psychotic prodrome presentation is relatively well-documented in the literature. The prodrome often includes delusions and hallucinations, which are severe, progress quickly, and are medication resistant. In addition to the widely recognized positive psychotic symptom prodrome, a negative-dominant prodrome including depressive symptoms may occur more commonly in ANMDARE than is widely acknowledged, and clinicians should be attentive to unexpected, atypical emergence of depressive symptoms in their patients as a possible sign of early ANMDARE.
Submitted: November 5, 2019; accepted February 3, 2020.
Published online: June 18, 2020.
Disclosure of off-label usage: The authors have determined that, to the best of their knowledge, no investigational information about pharmaceutical agents or device therapies that is outside US Food and Drug Administration-approved labeling has been presented in this article.
Financial disclosure: Drs Nguyen, Young, and Bourgeois have no personal affiliations or financial relationships with any commercial interest to disclose relative to the article.
Funding/support: None.
REFERENCES
1.Moscato EH, Peng X, Jain A, et al. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2014;76(1):108-119. PubMed CrossRef
2.Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr Res. 2016;176(1):36-40. PubMed CrossRef
3.Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol Rev. 2017;97(2):839-887. PubMed CrossRef
4.Zandi MS, Irani SR, Lang B, et al. Disease-relevant autoantibodies in first episode schizophrenia. J Neurol. 2011;258(4):686-688. PubMed CrossRef
5.Lehmann-Facius H. לber die Liquordiagnose der schizophrenien. Klin Wochenschr. 1937;16:1646-1648. CrossRef
6.Leypoldt F, Wandinger KP, Bien CG, et al. Autoimmune encephalitis. Eur Neurol Rev. 2013;8(1):31-37. PubMed CrossRef
7.Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol. 2001;50(1):73-78. PubMed CrossRef
8.Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005;128(pt 8):1764-1777. PubMed CrossRef
9.Dalmau J, T×¼z×¼n E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36. PubMed CrossRef
10.Castillo-Gomez E, Kästner A, Steiner J, et al. The brain as immunoprecipitator of serum autoantibodies against N-methyl-D-aspartate receptor subunit NR1. Ann Neurol. 2016;79(1):144-151. PubMed CrossRef
11.Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis in psychiatry. Curr Psychiatry Rev. 2011;7(3):189-193. PubMed CrossRef
12.Reddy MSS, Thippeswamy H, Ganjekar S, et al. Anti-NMDA receptor encephalitis presenting as postpartum psychosis: a clinical description and review. Arch Women Ment Health. 2018;21(4):465-469. PubMed CrossRef
13.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-1098. PubMed CrossRef
14.Moura M, Silva-Dos-Santos A, Afonso J, et al. First-episode psychosis in a 15 year-old female with clinical presentation of anti-NMDA receptor encephalitis: a case report and review of the literature. BMC Res Notes. 2016;9(1):374. PubMed CrossRef
15.Herken J, Pr×¼ss H. Red flags: clinical signs for identifying autoimmune encephalitis in psychiatric patients. Front Psychiatry. 2017;8:25. PubMed CrossRef
16.Al-Diwani A, Handel A, Townsend L, et al. The psychopathology of NMDAR-antibody encephalitis in adults: a systematic review and phenotypic analysis of individual patient data. Lancet Psychiatry. 2019;6(3):235-246. PubMed CrossRef
17.Kayser MS, Titulaer MJ, Gresa-Arribas N, et al. Frequency and characteristics of isolated psychiatric episodes in anti-N-methyl-D-aspartate receptor encephalitis. JAMA Neurol. 2013;70(9):1133-1139. PubMed CrossRef
18.Conroy MA, Finch T, Levin TT, et al. Chronic schizophrenia later diagnosed with anti-NMDA receptor encephalitis: case report and review of the literature. Clin Schizophr Relat Psychoses. 2018;11(4):201-204. PubMed CrossRef
19.Mesquita J, Siva L. Anti-NMDA receptor encephalitis suspected as cause of drug-induced psychosis. J Neuropsychiatry Clin Neurosci. 2011;23(4):E2. PubMed CrossRef
20.Tsutsui K, Kanbayashi T, Tanaka K, et al. Anti-NMDA-receptor antibody detected in encephalitis, schizophrenia, and narcolepsy with psychotic features. BMC Psychiatry. 2012;12(1):37. PubMed CrossRef
21.Dahm L, Ott C, Steiner J, et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol. 2014;76(1):82-94. PubMed CrossRef
22.Hammer C, Stepniak B, Schneider A, et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry. 2014;19(10):1143-1149. PubMed CrossRef
23.Blinder T, Lewerenz J. Cerebrospinal fluid findings in patients with autoimmune encephalitis-a systematic analysis. Front Neurol. 2019;10:804. PubMed CrossRef
24.Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-177. PubMed CrossRef
25.Knudtzen FC, Nilsson AC, Skarphedinsson S, et al. False-positive anti-NMDA receptor antibodies in severe case of Lyme neuroborreliosis. Neurol Sci. 2020;41(1):197-199. PubMed CrossRef
26.Balu DT. The NMDA receptor and schizophrenia: from pathophysiology to treatment. Adv Pharmacol. 2016;76:351-382. PubMed CrossRef
27.Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33(6):523-533. PubMed CrossRef
28.Iizuka T, Sakai F. Anti-nMDA receptor encephalitis: clinical manifestations and pathophysiology [in Japanese]. Brain Nerve. 2008;60(9):1047-1060. PubMed
29.Gleichman AJ, Spruce LA, Dalmau J, et al. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci. 2012;32(32):11082-11094. PubMed CrossRef
30.Regan MC, Romero-Hernandez A, Furukawa H. A structural biology perspective on NMDA receptor pharmacology and function. Curr Opin Struct Biol. 2015;33:68-75. PubMed CrossRef
31.Karakas E, Simorowski N, Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009;28(24):3910-3920. PubMed CrossRef
32.Chazot PL, Cik M, Stephenson FA. An investigation into the role of N-glycosylation in the functional expression of a recombinant heteromeric NMDA receptor. Mol Membr Biol. 1995;12(4):331-337. PubMed CrossRef
33.Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. 2010;30(17):5866-5875. PubMed CrossRef
34.Akg×¼l G, McBain CJ. Diverse roles for ionotropic glutamate receptors on inhibitory interneurons in developing and adult brain. J Physiol. 2016;594(19):5471-5490. PubMed CrossRef
35.Manto M, Dalmau J, Didelot A, et al. In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J Rare Dis. 2010;5(1):31. PubMed CrossRef
36.Driesen NR, McCarthy G, Bhagwagar Z, et al. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol Psychiatry. 2013;18(11):1199-1204. PubMed CrossRef
37.Levite M. Glutamate receptor antibodies in neurological diseases: anti-AMPA-GluR3 antibodies, anti-NMDA-NR1 antibodies, anti-NMDA-NR2A/B antibodies, anti-mGluR1 antibodies or anti-mGluR5 antibodies are present in subpopulations of patients with either: epilepsy, encephalitis, cerebellar ataxia, systemic lupus erythematosus (SLE) and neuropsychiatric SLE, Sjogren’s syndrome, schizophrenia, mania or stroke. These autoimmune anti-glutamate receptor antibodies can bind neurons in few brain regions, activate glutamate receptors, decrease glutamate receptor’s expression, impair glutamate-induced signaling and function, activate blood brain barrier endothelial cells, kill neurons, damage the brain, induce behavioral/psychiatric/cognitive abnormalities and ataxia in animal models, and can be removed or silenced in some patients by immunotherapy. J Neural Transm (Vienna). 2014;121(8):1029-1075. PubMed CrossRef
38.Ankarcrona M, Dypbukt JM, Bonfoco E, et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15(4):961-973. PubMed CrossRef
39.T×¼z×¼n E, Zhou L, Baehring JM, et al. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol. 2009;118(6):737-743. PubMed CrossRef
40.Nauen DW. Extra-central nervous system target for assessment and treatment in refractory anti-N-methyl-D-aspartate receptor encephalitis. J Crit Care. 2017;37:234-236. PubMed CrossRef
41.Pillai SC, Gill D, Webster R, et al. Cortical hypometabolism demonstrated by PET in relapsing NMDA receptor encephalitis. Pediatr Neurol. 2010;43(3):217-220. PubMed CrossRef
42.Jones KC, Benseler SM, Moharir M. Anti-NMDA Receptor Encephalitis. Neuroimaging Clin N Am. 2013;23(2):309-320. PubMed CrossRef
43.Bobilev AM, Perez JM, Tamminga CA. Molecular alterations in the medial temporal lobe in schizophrenia [published online ahead of print June 18, 2019]. Schizophr Res. PubMed 202 2020;217:71Cros-85.
44.Melzer N, Budde T, Stork O, et al. Limbic encephalitis: potential impact of adaptive autoimmune inflammation on neuronal circuits of the amygdala. Front Neurol. 2015;6:171. PubMed CrossRef
45.Buzsסki G, Wang XJ. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35(1):203-225. PubMed CrossRef
46.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157-165. PubMed CrossRef
47.Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, et al. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology. 2011;77(6):589-593. PubMed CrossRef
48.Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain. 2010;133(pt 6):1655-1667. PubMed CrossRef
49.Phillips OR, Joshi SH, Narr KL, et al. Superficial white matter damage in anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry. 2018;89(5):518-525. PubMed CrossRef
50.Filatenkov A, Richardson TE, Daoud E, et al. Persistence of parenchymal and perivascular T-cells in treatment-refractory anti-N-methyl-D-aspartate receptor encephalitis. Neuroreport. 2017;28(14):890-895. PubMed CrossRef
51.Dickerson F, Stallings C, Vaughan C, et al. Antibodies to the glutamate receptor in mania. Bipolar Disord. 2012;14(5):547-553. PubMed CrossRef
52.Planagum× J, Leypoldt F, Mannara F, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behavior in mice. Brain. 2015;138(pt 1):94-109. PubMed CrossRef
53.Armangue T, Leypoldt F, Mסlaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol. 2014;75(2):317-323. PubMed CrossRef
54.Armangue T, Moris G, Cantar×n-Extremera V, et al; Spanish Prospective Multicentric Study of Autoimmunity in Herpes Simplex Encephalitis. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology. 2015;85(20):1736-1743. PubMed CrossRef
55.Gable MS, Gavali S, Radner A, et al. Anti-NMDA receptor encephalitis: report of ten cases and comparison with viral encephalitis. Eur J Clin Microbiol Infect Dis. 2009;28(12):1421-1429. PubMed CrossRef
56.Linnoila J, Pulli B, Armangué T, et al. Mouse model of anti-NMDA receptor post-herpes simplex encephalitis. Neurol Neuroimmunol Neuroinflamm. 2018;6(2):e529. PubMed CrossRef
57.Kothur K, Gill D, Wong M, et al. Cerebrospinal fluid cyto-/chemokine profile during acute herpes simplex virus induced anti-N-methyl-D-aspartate receptor encephalitis and in chronic neurological sequelae. Dev Med Child Neurol. 2017;59(8):806-814. PubMed CrossRef
58.Gibson LL, Pollak TA, Blackman G, et al. The psychiatric phenotype of anti-NMDA receptor encephalitis. J Neuropsychiatry Clin Neurosci. 2019;31(1):70-79. PubMed CrossRef
59.Lebon S, Mayor-Dubois C, Popea I, et al. Anti-N-methyl-D-aspartate (NMDA) receptor encephalitis mimicking a primary psychiatric disorder in an adolescent. J Child Neurol. 2012;27(12):1607-1610. PubMed CrossRef
60.Mantere O, Saarela M, Kieseppä T, et al. Anti-neuronal anti-bodies in patients with early psychosis. Schizophr Res. 2018;192:404-407. PubMed CrossRef
61.Sacré K, Lidove O, Chanson N, et al. Acute psychosis in anti-NMDA-receptor encephalitis. Presse Med. 2011;40(9 pt 1):882-884. PubMed CrossRef
62.Smith K. Mental health: a world of depression. Nature. 2014;515(7526):181. PubMed CrossRef
63.Warren N, O’ Gorman C, McKeon G, et al. Psychiatric management of anti-NMDAR encephalitis: a cohort analysis [published online ahead of print November 19, 2019]. Psychol Med. 2019;1-6. PubMed CrossRef
64.Gao HX, Sanders E, Tieng AT, et al. Sex and autoantibody titers determine the development of neuropsychiatric manifestations in lupus-prone mice. J Neuroimmunol. 2010;229(1-2):112-122. PubMed CrossRef
65.W×¼rdemann T, Kersten M, Tokay T, et al. Stereotactic injection of cerebrospinal fluid from anti-NMDA receptor encephalitis into rat dentate gyrus impairs NMDA receptor function. Brain Res. 2016;1633:10-18. PubMed CrossRef
66.Omdal R, Brokstad K, Waterloo K, et al. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur J Neurol. 2005;12(5):392-398. PubMed CrossRef
67.Lapteva L, Nowak M, Yarboro CH, et al. Anti-N-methyl-D-aspartate receptor antibodies, cognitive dysfunction, and depression in systemic lupus erythematosus. Arthritis Rheum. 2006;54(8):2505-2514. PubMed CrossRef
68.Lauvsnes MB, Maroni SS, Appenzeller S, et al. Memory dysfunction in primary Sjögren’s syndrome is associated with anti-NR2 antibodies. Arthritis Rheum. 2013;65(12):3209-3217. PubMed CrossRef
69.Gariup M, Lera-Miguel S, Torres F, et al. Autoantibodies, elevated cytokines, and neurocognitive abnormalities in offspring of women with systemic lupus erythematosus: comparison with healthy controls. Clin Rheumatol. 2019;38(9):2529-2539. PubMed CrossRef
70.Sveinsson O, Granqvist M, Forslin Y, et al. Successful combined targeting of B- and plasma cells in treatment refractory anti-NMDAR encephalitis. J Neuroimmunol. 2017;312:15-18. PubMed CrossRef
71.Fawcett RG. Acute psychosis associated with anti-NMDA-receptor antibodies and bilateral ovarian teratomas: a case report. J Clin Psychiatry. 2010;71(4):504. PubMed CrossRef
72.Florance NR, Davis RL, Lam C, et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009;66(1):11-18. PubMed CrossRef
73.Chapman MR, Vause HE. Anti-NMDA receptor encephalitis: diagnosis, psychiatric presentation, and treatment. Am J Psychiatry. 2011;168(3):245-251. PubMed CrossRef
Enjoy this premium PDF as part of your membership benefits!



