Objective: Pimavanserin is a 5-hydroxytryptamine-2A antagonist and inverse receptor agonist. This phase 2 study examined the efficacy and safety of pimavanserin as adjunctive therapy in patients with major depressive disorder (MDD).
Methods: This was a multicenter, randomized, double-blind, placebo-controlled study in patients with DSM-5-defined MDD and an inadequate response to a selective serotonin reuptake inhibitor (SSRI) or serotonin-norepinephrine reuptake inhibitor (SNRI). Using a 2-stage sequential parallel-comparison design, patients were initially randomized in a 3:1 ratio to placebo or pimavanserin added to ongoing SSRI or SNRI therapy; at 5 weeks, placebo nonresponders were re-randomized to placebo or pimavanserin for an additional 5 weeks. Key endpoints were change from baseline to the end of each stage in 17-item Hamilton Depression Rating Scale (HDRS-17) total score and Sheehan Disability Scale (SDS) score.
Results: Between December 2016 and October 2018, 207 patients were randomized. For the prespecified pooled Sequential Parallel Comparison Design analyses of Stages 1 and 2, the least squares (LS) mean (SE) difference for the HDRS-17 total score was −1.7 (0.85) (P = .039) and for the SDS score was −0.8 (0.29) (P = .004). At week 5 of Stage 1, LS mean (SE) difference for pimavanserin versus placebo was significant for changes on the HDRS-17 (−4.0 [1.09], P = .0003) and SDS (−1.2 [0.40], P = .0036) with effect sizes of 0.626 and 0.498, respectively. Early and sustained separation of pimavanserin from placebo (P < .05) occurred at 1 week. The most common adverse events with pimavanserin were dry mouth, nausea, and headache.
Conclusions: Pimavanserin demonstrated robust efficacy in patients with MDD and an inadequate response to an SSRI or SNRI. Tolerability was consistent with previous experience.
Trial Registration: ClinicalTrials.gov identifier: NCT03018340
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
ABSTRACT
Objective: Pimavanserin is a 5-hydroxytryptamine-2A antagonist and inverse receptor agonist. This phase 2 study examined the efficacy and safety of pimavanserin as adjunctive therapy in patients with major depressive disorder (MDD).
Methods: This was a multicenter, randomized, double-blind, placebo-controlled study in patients with DSM-5-defined MDD and an inadequate response to a selective serotonin reuptake inhibitor (SSRI) or serotonin-norepinephrine reuptake inhibitor (SNRI). Using a 2-stage sequential parallel-comparison design, patients were initially randomized in a 3:1 ratio to placebo or pimavanserin added to ongoing SSRI or SNRI therapy; at 5 weeks, placebo nonresponders were re-randomized to placebo or pimavanserin for an additional 5 weeks. Key endpoints were change from baseline to the end of each stage in 17-item Hamilton Depression Rating Scale (HDRS-17) total score and Sheehan Disability Scale (SDS) score.
Results: Between December 2016 and October 2018, 207 patients were randomized. For the prespecified pooled Sequential Parallel Comparison Design analyses of Stages 1 and 2, the least squares (LS) mean (SE) difference for the HDRS-17 total score was −1.7 (0.85) (P = .039) and for the SDS score was −0.8 (0.29) (P = .004). At week 5 of Stage 1, LS mean (SE) difference for pimavanserin versus placebo was significant for changes on the HDRS-17 (−4.0 [1.09], P = .0003) and SDS (−1.2 [0.40], P = .0036) with effect sizes of 0.626 and 0.498, respectively. Early and sustained separation of pimavanserin from placebo (P < .05) occurred at 1 week. The most common adverse events with pimavanserin were dry mouth, nausea, and headache.
Conclusions: Pimavanserin demonstrated robust efficacy in patients with MDD and an inadequate response to an SSRI or SNRI. Tolerability was consistent with previous experience.
Trial Registration: ClinicalTrials.gov identifier: NCT03018340
J Clin Psychiatry 2019;80(6):19m12928
To cite: Fava M, Dirks B, Freeman MP, et al. A phase 2, randomized, double-blind, placebo-controlled study of adjunctive pimavanserin in patients with major depressive disorder and an inadequate response to therapy (CLARITY). J Clin Psychiatry. 2019;80(6):19m12928.
To share: https://doi.org/10.4088/JCP.19m12928
© Copyright 2019 Physicians Postgraduate Press, Inc.
aDepartment of Psychiatry, Massachusetts General Hospital, and Harvard Medical School, Boston, Massachusetts
bACADIA Pharmaceuticals Inc, San Diego, California
cDepartment of Psychiatry and Behavioral Neurobiology, University of Alabama at Birmingham, Birmingham, Alabama
dDepartment of Psychiatry, Perelman School of Medicine, University of Pennsylvania and the Philadelphia Veterans Affairs Medical Center, Philadelphia, Pennsylvania
eDepartment of Psychiatry, University of Texas Southwestern Medical Center, Dallas, Texas
*Corresponding author: Maurizio Fava, MD, Department of Psychiatry, Harvard Medical School, 55 Fruit St, Bulfinch 351, Boston, MA 02114 ([email protected]).
Major depressive disorder (MDD) affects 300 million persons worldwide,1 causing substantial morbidity and mortality.2 Drug therapy is effective,3 but fewer than one-third of patients achieve remission.4-8 A few atypical antipsychotics are approved by the US Food and Drug Administration (FDA) for use in adjunctive treatment of patients with an inadequate response to antidepressants. Currently available atypical antipsychotics may reduce symptoms9; however, their use may be limited by associated weight gain, metabolic effects, and extrapyramidal effects.4,10-12 Thus, other options are needed for patients with MDD and inadequate treatment response.13
Pimavanserin, approved for treating Parkinson’s disease psychosis, is a potent 5-hydroxytryptamine-2A (5-HT2A) receptor antagonist/inverse agonist with lesser activity as a 5-HT2C antagonist/inverse agonist but no activity at adrenergic, dopaminergic, histaminergic, or muscarinic receptors.14 Activity of some antidepressant drugs has been attributed to blockade of the 5-HT2A receptor.15 Mixed β-adrenoceptor/5-HT1A antagonists may enhance the clinical action of selective serotonin reuptake inhibitors (SSRI), activity demonstrated with drugs that combine 5-HT reuptake inhibition and partial agonism at 5-HT1A receptors.16 Pimavanserin demonstrates a receptor-binding profile similar to those of other drugs with antidepressant activity.17,18 This report presents data from a phase 2 study (CLARITY; ClinicalTrials.gov identifier: NCT03018340) undertaken to investigate the efficacy and tolerability of pimavanserin administered as adjunctive therapy for patients with MDD and an inadequate response to antidepressant therapy.
METHODS
The study protocol was reviewed by an independent ethics committee or institutional review board at each study site and implemented following the principles of Good Clinical Practice derived from the Declaration of Helsinki and in accordance with local regulations and International Council of Harmonization guidelines. All patients provided written informed consent prior to any study procedures.
Study Design
This was a multicenter, randomized, double-blind, placebo-controlled study in patients with MDD and inadequate responses to treatment with an SSRI or serotonin-norepinephrine reuptake inhibitor (SNRI). The primary objective was to evaluate efficacy, and secondary objectives were to evaluate safety and tolerability, effects on disability, clinician’s global assessment, patient-reported quality of life, perception of treatment, sleepiness, sexual functioning, impulsivity, and irritability of pimavanserin in this patient population. The study consisted of an 8- to 21-day screening period, a 10-week double-blind treatment period, and a 30-day safety follow-up period. To ensure that appropriate patients were enrolled, a remote interview was conducted by Massachusetts General Hospital Clinical Trials Network and Institute raters between the screening and baseline visits. Screening assessments consisted of the SAFER Interview,19 which included the Montgomery-Asberg Depression Rating Scale (MADRS),20 the Clinical Global Impression-Severity of Illness scale (CGI-S),21 and the Massachusetts General Hospital Antidepressant Treatment Questionnaire (MGH ATRQ).22
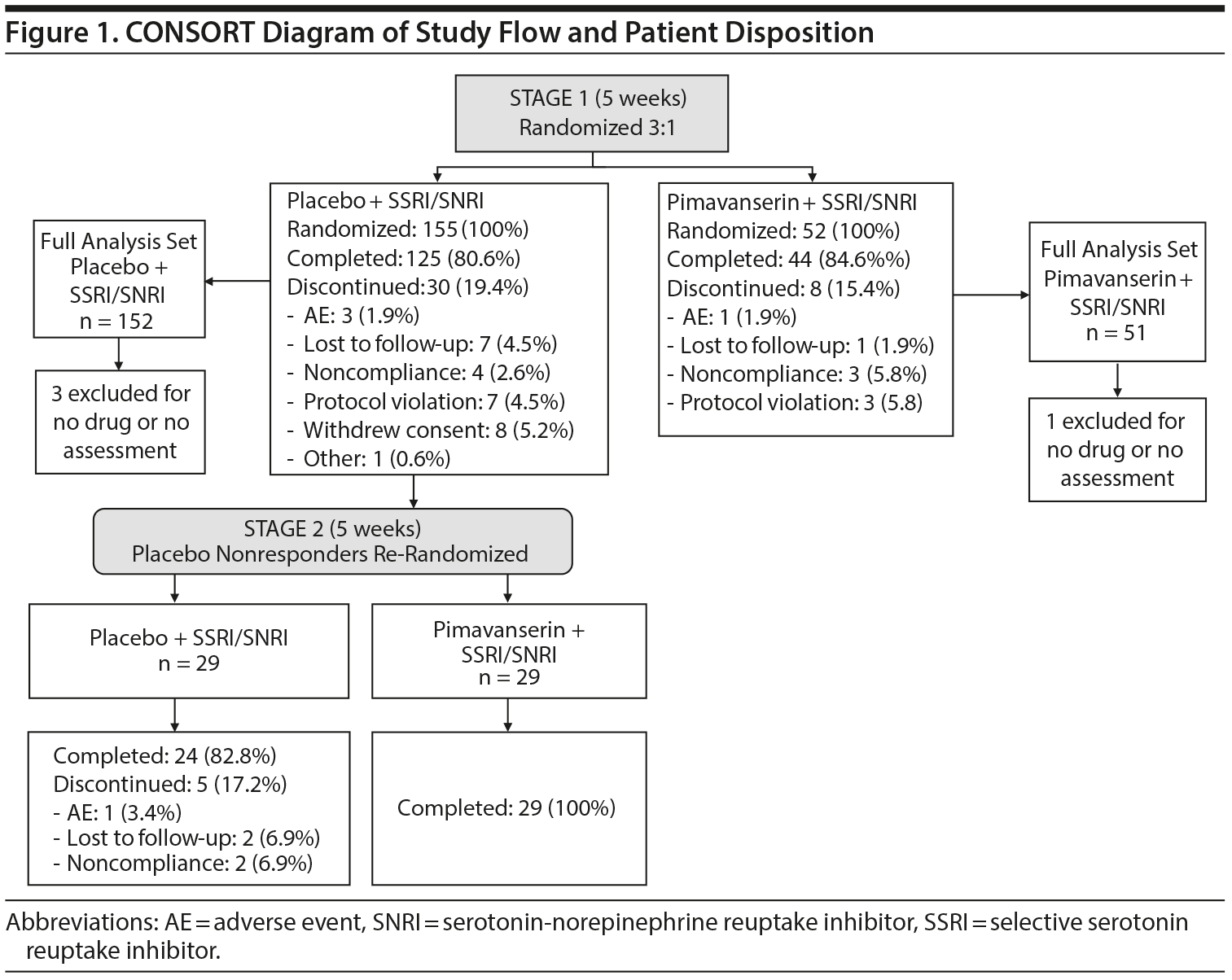
The study utilized a 2-stage Sequential Parallel-Comparison Design (SPCD)23 whereby, following screening, eligible patients were randomized in Stage 1 in a 3:1 ratio to placebo or pimavanserin added to their current SSRI or SNRI therapy for 5 weeks. At the end of 5 weeks, placebo nonresponders (17-item Hamilton Depression Rating Scale [HDRS-17]24 total score > 14 and < 50% reduction in score from baseline) were re-randomized to placebo or pimavanserin (1:1 ratio) added to current therapy for an additional 5 weeks. All patients assigned to pimavanserin in Stage 1 continued treatment with pimavanserin in Stage 2, whereas responders to placebo in Stage 1 remained on placebo in Stage 2. Patients were randomly assigned via an interactive voice response system to pimavanserin 34 mg once daily or placebo. All patients continued on their SSRI or SNRI at a stable dose for the duration of the study. Patients and investigators and their staff were blinded to treatment assignment.
Patient Selection
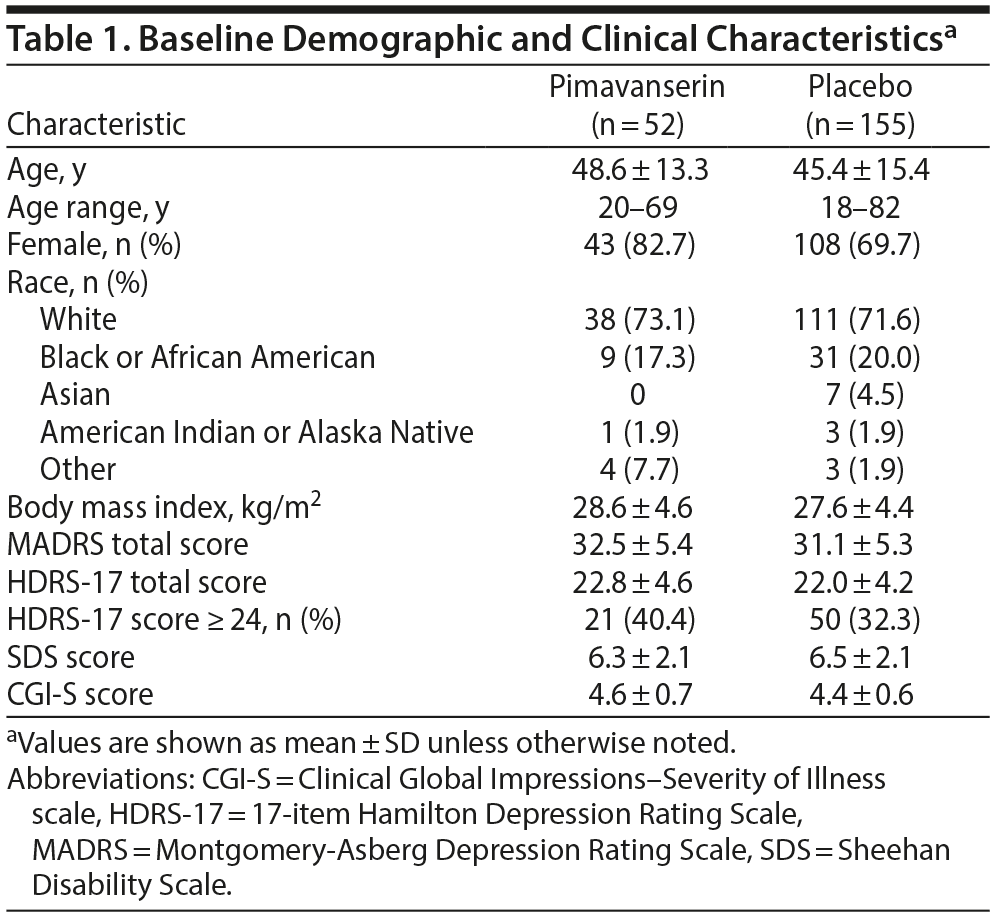
Male or female patients ≥ 18 years of age with a body mass index (BMI) between 19 and 35 kg/m2 inclusive were eligible if they had a primary diagnosis of MDD and a current major depressive episode (MDE) defined by the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), and confirmed by the Structured Clinical Interview for DSM-5, Clinical Trials Version (SCID-5-CT).25 Eligible patients had a history of MDD for ≥ 1 year prior to screening, a MADRS total score > 20, and a CGI-S score ≥ 4 (moderately ill or worse) at both screening and baseline. Eligible patients also had a history of inadequate response to 1 or 2 antidepressant treatments during the current depression episode. Inadequate treatment response was determined with the MGH ATRQ, administered during the SAFER interview. Eligible patients were receiving treatment for their current episode with exactly 1 of the following drugs at approved doses: citalopram, desvenlafaxine, duloxetine, escitalopram, fluoxetine, paroxetine, sertraline, venlafaxine, or venlafaxine extended-release. The minimum trial duration for inclusion was 8 weeks (with the last 4 weeks on a stable dose).
Eligible women were of non-childbearing potential or agreed to use 2 acceptable methods of contraception throughout the study. A negative serum pregnancy test at screening and urine pregnancy test at baseline were required for inclusion. Patients were excluded for any medical or psychiatric condition or clinically significant laboratory abnormality that could interfere with safety or the conduct of the study. Patients who were actively suicidal or had attempted suicide within the past 2 years were excluded. Complete selection criteria are detailed in Supplementary Table 1.
Click figure to enlarge
Study Assessments
Clinic visits occurred weekly from weeks 1 through 10 (end of study). The MADRS was administered at screening and baseline. The HDRS-17, Sheehan Disability Scale (SDS),26 CGI-S, and Karolinska Sleepiness Scale (KSS)27 were administered at baseline and weekly during the study. The 12-Item Short-Form Health Survey (SF-12),28 Drug Attitude Inventory (DAI-10),29 and MGH Sexual Functioning Index (MGH-SFI)30 were administered at baseline and weeks 5 and 10. The Barratt Impulsiveness Scale (BIS-11)31 and Sheehan Irritability Scale (SIS)32 were administered at baseline and weeks 1, 3, 5, 6, 8, and 10. Safety assessments, including adverse events (AEs), physical examination, vital signs, 12-lead electrocardiogram (ECG), and clinical laboratory tests (hematology, chemistry, urinalysis, prolactin), were performed at baseline and routinely during the study. The Columbia-Suicide Severity Rating Scale (C-SSRS)33 was administered at baseline and weekly, and the Barnes Akathisia Rating Scale (BARS),34 Abnormal Involuntary Movement Scale (AIMS),35 and Simpson-Angus Scale (SAS)36 were administered at baseline and weeks 1, 5, 6, and 10. Treatment response was defined as a ≥ 50% reduction from baseline for the HDRS-17 total score, and remission was defined as a HDRS-17 total score ≤ 7.
Statistical Analyses
- An inadequate response to antidepressant treatment is a common cause of poor outcomes in patients with major depressive disorder (MDD), and currently available adjunctive treatments are not adequate.
- For patients with MDD not responding to antidepressant treatment, adjunctive pimavanserin produced a robust treatment response.
- Additional clinical studies with adjunctive pimavanserin are planned in patients with MDD.
A total sample size of 168 evaluable patients was estimated to provide at least 80% power at a 2-sided significance level of .05. Adjusting for a potential non-evaluable rate of up to 10%, approximately 188 patients were planned for randomization. The primary efficacy endpoint, change from baseline to the end of each stage for the HDRS-17 total score, was evaluated using mixed models for repeated measures (MMRM) with effects for treatment group, visit, treatment-by-visit interaction, baseline HDRS-17 total score, and the baseline HDRS-17 total score-by-visit interaction. An unstructured covariance matrix was used, and the Kenward-Roger approximation was used to adjust the denominator degrees of freedom. The treatment effect was assessed as the treatment difference in least squares mean (LS mean) change from baseline to the end of each stage and the corresponding 95% CIs. Cohen d effect size was calculated for comparisons between treatments. Similar statistical methods were used to analyze other continuous endpoints. For the CGI-Improvement scale (CGI-I), baseline CGI-S score was used as the covariate. For the SF-12, DAI-10, and MGH-SFI, which were assessed only at baseline and at the end of the efficacy period, analysis of covariance with effects for treatment group and baseline score was used instead of the MMRM model. For response and remission rates, treatment groups were compared using the Pearson χ2 test. Overall treatment effects were assessed as the weighted treatment differences in LS mean change for the 2 stages. The number needed to treat (NNT) was calculated for response and remission from the absolute rate of improvement at week 5. Efficacy data were analyzed for the full analysis set (FAS) for each of the 2 stages, comprising all randomized patients who received ≥ 1 dose of blinded study drug and who had a baseline value and at least 1 post-baseline value for the HDRS-17 total score within each stage. The safety analysis set included all patients receiving ≥ 1 dose of blinded study drug for each of the 2 stages.
RESULTS
Between December 2016 and October 2018, 207 patients were randomized at 27 study sites and included in the safety population (Figure 1). In Stage 1, 152 (98.1%) and 51 (98.1%) patients in the placebo and pimavanserin groups, respectively, were included in the FAS population; 4 patients were excluded because they did not receive study drug or had no baseline and posttreatment HDRS-17 score. In Stage 2, 29 patients in both placebo and pimavanserin groups were included for the safety and FAS populations.
Click figure to enlarge
At baseline, treatment groups were generally comparable for demographic and clinical characteristics (Table 1). Patients in the pimavanserin group tended to have more severe depression based on a greater proportion with a HDRS-17 total score ≥ 24 (40.4% vs 32.3%) and a greater proportion with a score of 5 (markedly ill) or 6 (severely ill) on the CGI-S (48.1% vs 38.7%).
Efficacy
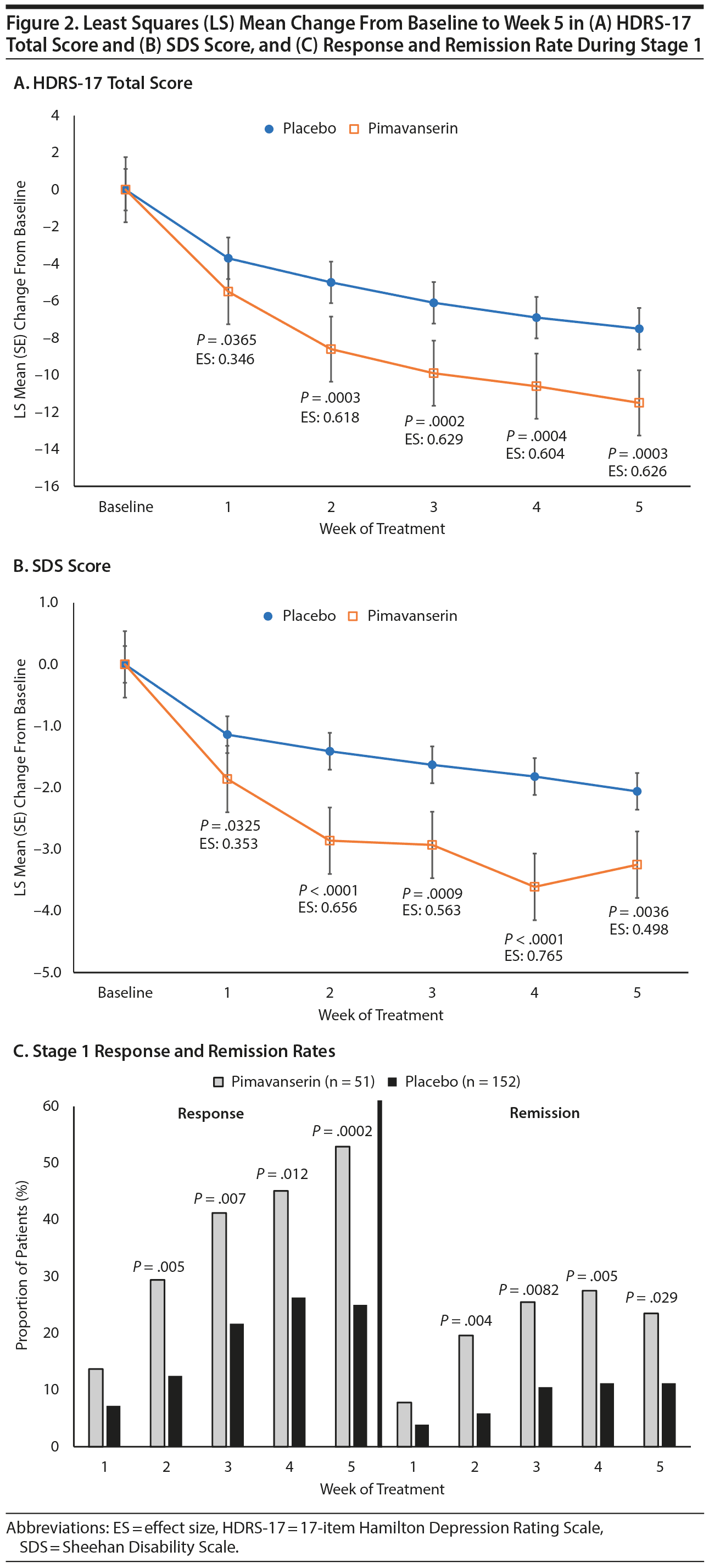
For the prespecified pooled SPCD analyses of Stages 1 and 2, a significantly greater improvement was observed with pimavanserin than placebo for the HDRS-17 total score (LS mean difference [SE] = −1.7 [0.85], P = .039) and SDS score (LS mean difference = −0.8 [0.29], P = .004). At week 5 of Stage 1, LS mean (SE) change from baseline for the HDRS-17 was −11.5 (0.94) for pimavanserin and −7.5 (0.55) for placebo (LS mean difference = −4.0 [1.09], P = .0003; effect size: 0.626), and for SDS, LS mean change was −3.3 (0.35) for pimavanserin and −2.1 (0.20) for placebo (LS mean difference = −1.2 [0.40], P = .0036, effect size: 0.498). LS mean change from baseline was significantly (P < .05) greater for pimavanserin versus placebo from week 1 to week 5 for both the HDRS-17 and SDS (Figure 2). During Stage 1, response and remission rates were significantly (P < .05) greater with pimavanserin versus placebo from week 2 through week 5 (Figure 2). The NNT for response and remission in Stage 1 was 3.6 and 8.1, respectively. In Stage 2, the difference between pimavanserin and placebo was not significant for the HDRS-17 (delta = 0.5; P = .6940; Cohen d = -0.107).
Click figure to enlarge
When data were stratified by baseline HDRS-17 total score < 24 or ≥ 24, a more robust treatment effect was observed for both the HDRS-17 and SDS in the subgroup with more severe baseline depression (Supplementary Table 2). For the combined Stages 1 and 2, LS mean difference in the severe subgroup was −3.6 (1.4) for the HDRS-17 (P = .011) and −1.14 (0.47) for the SDS (P = .014). At week 5 in Stage 1, LS mean difference in the severe subgroup was −7.0 (2.1) for the HDRS-17 (P = .0014; effect size: 0.933) and −2.0 (0.68) for the SDS (P = .0054; effect size: 0.815). No significant differences were observed for HDRS-17 or SDS during Stage 2.
Click figure to enlarge
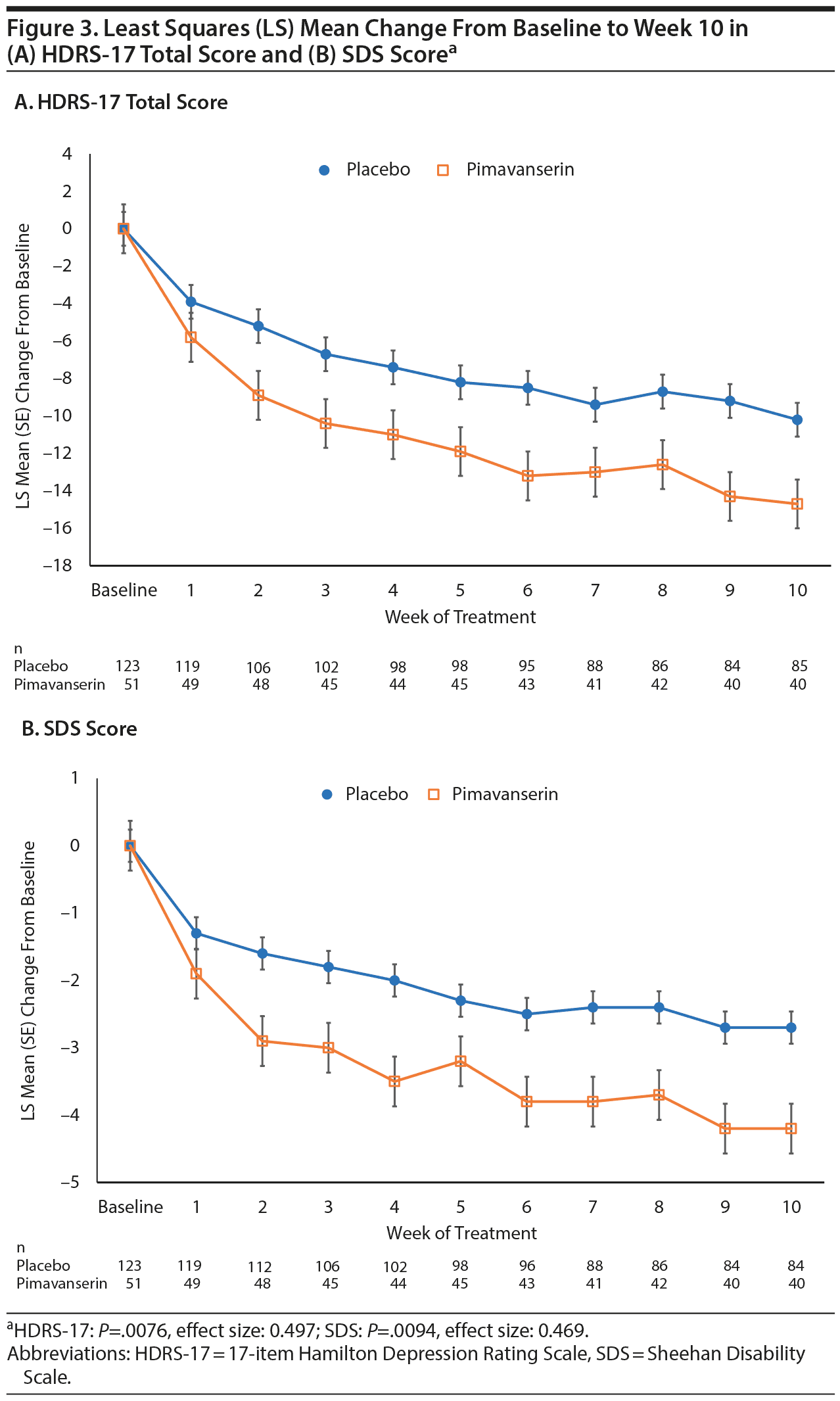
In the prespecified subgroup analyses by background antidepressant, significant differences from placebo (P < .05) were observed both for patients receiving an SSRI and for patients receiving SNRI as a background therapy from weeks 2 to week 5 in Stage 1 and for the overall treatment effect (P = .0193) with effect sizes of 0.43 to 0.61, respectively (data not shown). In a prespecified analysis of patients receiving continuous treatment with pimavanserin or placebo for 10 weeks in Stages 1 and 2, the treatment difference in LS mean change from baseline to week 10 was significant for both the HDRS-17 (P = .0076; effect size: 0.497) and the SDS (P = .0094; effect size: 0.469) in favor of pimavanserin (Figure 3).
Click figure to enlarge
For secondary efficacy endpoints in Stage 1, significant LS mean differences in favor of pimavanserin were observed at week 5 for the CGI-S (P = .0001), CGI-I (P = .001) (Supplementary Table 2).
Safety and Tolerability
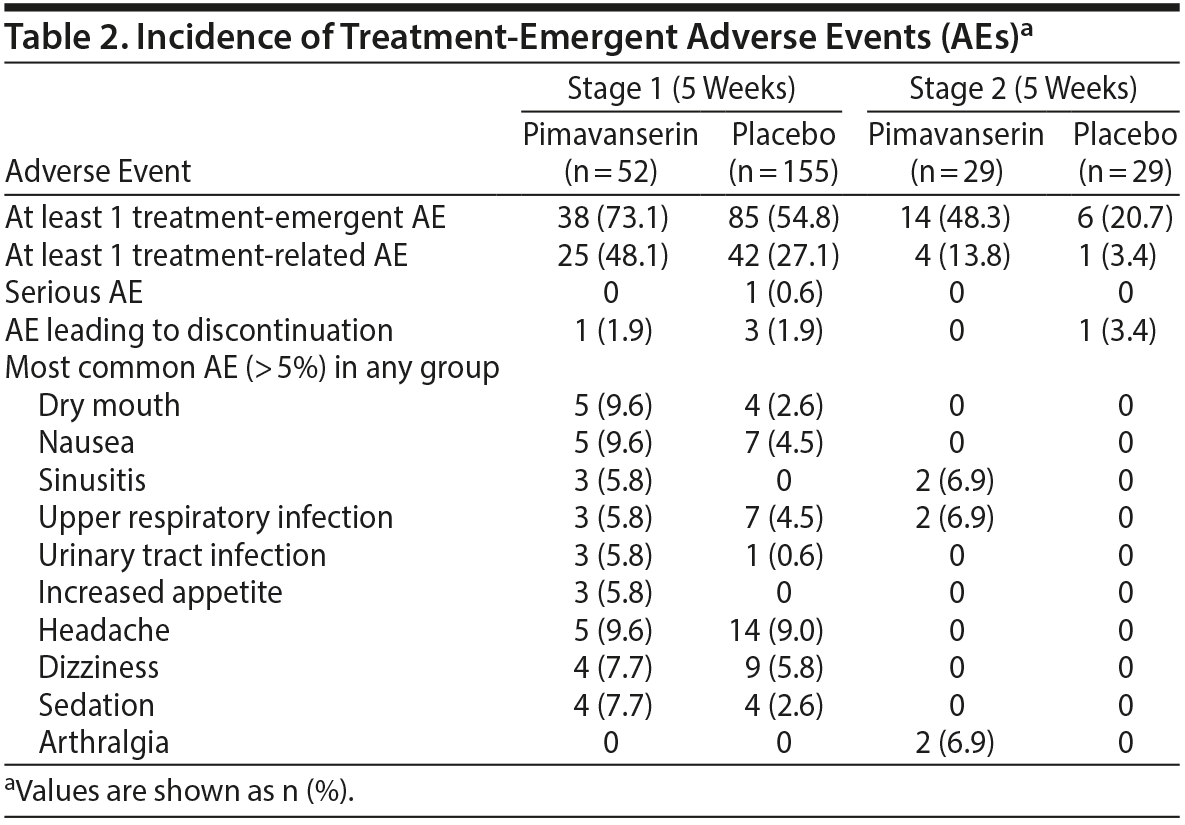
Treatment-related AEs occurred in 27.1% and 48.1% of patients in the placebo and pimavanserin groups, respectively, during Stage 1 and in 3.4% and 13.8% of patients in the placebo and pimavanserin groups, respectively, during Stage 2 (Table 2). The most common AEs in the pimavanserin group were dry mouth, nausea, and headache, all with frequency of less than 10%. Two serious AEs (bladder stones, prostate cancer) occurred in the placebo group and 1 (acute myocardial infarction) in the pimavanserin group; all were considered unrelated to therapy, and patients remained on study drug. During Stage 1, 3 patients (1.9%) discontinued the study due to an AE in the placebo group and 1 (1.9%) in the pimavanserin group. Additionally, 1 patient (3.4%) discontinued placebo during Stage 2 (Figure 1). No deaths occurred, and no clinically relevant changes in vital signs, clinical laboratory testing, or ECG findings were observed. Mean changes from baseline for plasma glucose and lipid parameters were minimal and not clinically significant in either treatment group (Supplementary Table 3). At week 5, the mean change from baseline for serum prolactin levels was 10.2 (9.5) μIU/mL for placebo and −28.4 (15.6) μIU/mL for pimavanserin (Supplementary Table 3). A low rate of extrapyramidal symptoms was observed; 2 events (1.3%) of akathisia occurred with placebo during Stage 1; 1 event (3.4%) of bradykinesia and 1 (3.4%) of cogwheel rigidity occurred with pimavanserin occurred during Stage 2.
For the MGH-SFI at week 5 of Stage 1, the LS mean difference from baseline for pimavanserin versus placebo was −0.6 (0.17) (P = .0002; effect size: 0.614), indicating superior sexual functioning among pimavanserin-treated patients. For the KSS at week 5 of Stage 1, LS mean difference from baseline for pimavanserin versus placebo was −1.1 (0.30) (P = .0003; effect size: 0.627), indicating less somnolence among pimavanserin-treated subjects (Supplementary Table 2). In Stage 1, no difference from baseline to week 5 for the BIS-11 score was observed for pimavanserin versus placebo (Supplementary Table 2). In Stage 2 and the overall weighted analysis of Stages 1 and 2 for the BIS-11, a significant improvement from baseline was observed with pimavanserin versus placebo. In Stage 1, a significant improvement for the SIS from baseline to week 5 was observed with pimavanserin versus placebo (Supplementary Table 2). In Stage 2 and overall, no difference from baseline to week 5 in SIS score was observed between pimavanserin and placebo.
DISCUSSION
The results of this phase 2 study overall showed statistically significant and clinically relevant improvements in the HDRS-17 total score and SDS score with pimavanserin. Particularly robust efficacy was observed during the all-inclusive Stage 1 of the study in which all initially randomized patients are analyzed. The observed effect sizes for measures of depressive symptoms and function at week 5 of Stage 1 were in the range of 0.6 and 0.5, respectively. Additionally, in this study, pimavanserin has demonstrated early separation from placebo at week 1 during Stage 1 for both the HDRS-17 and the SDS. Moreover, significant improvements were observed for a number of secondary endpoints, including sexual functioning, daytime sleepiness, and mental health-related quality of life. The safety and tolerability profile of pimavanserin was consistent with the existing product labeling, and no new safety findings were reported.
One challenge when conducting randomized controlled trials in MDD is the unpredictable and often substantial placebo response, which can undermine the ability to detect a statistically significant difference between drug and placebo groups.37-39 The SPCD was developed as a clinical trial methodology to mitigate risk in such instances, reducing the effect of placebo in Stage 2 with the use of a double-blind antidepressant-placebo lead-in in Stage 1.23 The SPCD is especially useful in drug development as a method to reduce sample size, provide a consistent pool of patients, and reduce variability. Given that it is very challenging to identify depressed patients who are likely to respond to placebo,40 by identifying and removing placebo responders during Stage 1 with the SPCD methodology, treatment differences are typically expected to be more detectable in patients who continue into Stage 2 and are re-randomized to staying on placebo or go on active treatment. An additional advantage of the SPCD methodology is the fact that it offers “2 shots on goal” within the same trial to enhance the probability of detecting an effect. This was, in fact, the case in our study, in which the robustness of the effect detected in Stage 1 of SPCD led to overall efficacy despite the lack of effect in Stage 2. The latter was probably due to the markedly smaller than expected sample size of Stage 2 and to the fact that Stage 2 was probably enriched with refractory (and therefore less informative) depressed patients, as a result of the dual requirement for re-randomization.
In the present trial, significant differences were observed between pimavanserin and placebo for the prespecified pooled SPCD analyses for the HDRS-17 total score and the SDS score. When each stage is analyzed separately, particularly robust efficacy results with pimavanserin were observed in Stage 1 with significant mean change from baseline to week 5 for the HDRS-17 and SDS and substantial effect sizes of 0.626 and 0.498, respectively. In contrast, studies10,11 with atypical antipsychotics as adjunctive therapy in patients with MDD report effect sizes of 0.27 to 0.43 symptoms. However, because, unexpectedly, few placebo nonresponders—approximately half of the anticipated number—were re-randomized to Stage 2, no statistical separation was observed in this group of re-randomized patients. Additionally, relatively stringent re-randomization criteria may have led to inclusion of a group of subjects with less capacity to improve in Stage 2 and thus contributed to this observation.
As a further indication of robust benefit of pimavanserin adjunctive therapy, a prespecified analysis of 10-week efficacy for patients who received the same, originally assigned treatment through both stages of the study (n = 178, 126 placebo and 52 pimavanserin) showed statistically significant and clinically robust separation from placebo on the HDRS-17 and SDS with the effect sizes of 0.497 and 0.469, respectively. Significant improvement in function on the SDS suggests potentially broader beneficial effects of pimavanserin as adjunctive treatment. Few studies with atypical antipsychotics in MDD included assessment of functional improvement.
This study included the SDS as a key secondary endpoint to assess functional disability, and improvement in workplace function has been demonstrated with antidepressant therapy in patients with MDD.41 The SDS is well validated and widely accepted for assessing functional outcomes in patients with MDD.42 In a systematic review of studies that assessed functional outcomes, the authors suggested that improvements in function (SDS) should be considered for inclusion as co-endpoints with symptomatic assessments when evaluating treatments for MDD.43 Routine assessments of both symptoms and function could be helpful for minimizing residual effects that increase the risk for relapse or recurrence.43
Pimavanserin was well tolerated in this study. The AE profile was consistent with those of previous studies of pimavanserin for Parkinson’s disease psychosis and Alzheimer’s disease psychosis,44,45 and discontinuations for AEs were lower with pimavanserin than with placebo. Importantly, pimavanserin was associated with low rates of daytime sleepiness, weight gain, metabolic changes, and sexual dysfunction. In contrast, use of atypical antipsychotics may be limited by weight gain, metabolic disturbances (glucose intolerance, diabetes, lipid disorders, hyperprolactinemia), and daytime sleepiness.10,46-48 In addition, most conventional antidepressants are well known to cause sexual dysfunction in at least 50% of patients with MDD.49
Limitations of this study include a relatively short length of treatment as well as a small sample size, particularly in Stage 2. A longer duration of treatment in MDD may be necessary to more completely elucidate the full effect of drug treatment.50 However, this study was designed to establish an acute signal of efficacy for pimavanserin in an MDD population, and the SPCD design permitted use of a smaller sample size to ascertain efficacy.
Despite these limitations, pimavanserin demonstrated robust and clinically meaningful efficacy in MDD patients with inadequate response to antidepressant therapy. Importantly, pimavanserin was not associated with significant metabolic dysregulation, sexual dysfunction, or extrapyramidal symptoms. Thus, pimavanserin may offer an effective alternative as adjunctive therapy for patients with MDD and an inadequate response to antidepressant treatment without the safety and tolerability concerns of atypical antipsychotics. A phase 3 program of adjunctive pimavanserin in patients with MDD inadequately responsive to SSRI or SNRI therapy has been initiated.
Submitted: May 24, 2019; accepted August 19, 2019.
Published online: September 24, 2019.
Author contributions: All authors had full access to all of the data in the study and had full responsibility for the content of the manuscript for publication. The corresponding author was responsible for the final review and had final responsibility for the decision to submit for publication.
Potential conflicts of interest: Dr Freeman has received advisory/consulting fees from Alkermes, Otsuka, Janssen, Sage, JDS Therapeutics, and Sunovion; took part in an independent data safety and monitoring committee for Janssen (Johnson & Johnson); has performed medical editing for the Global Organization for EPA and DHA Omega-3 newsletter; has received research support through Massachusetts General Hospital (MGH) from National Pregnancy Registry for Atypical Antipsychotics, Alkermes, AstraZeneca, Otsuka, Forest/Actavis, Ortho-McNeil Janssen, and Sunovion; has received other research support from Takeda and JayMac; and as an employee of MGH, works with the MGH Clinical Trials Network and Institute, which has had research funding from multiple pharmaceutical companies and National Institute of Mental Health (NIMH). Dr Fava has received research support from Abbott, ACADIA, Alkermes, American Cyanamid, Aspect Medical Systems, AstraZeneca, Avanir, Axsome Therapeutics, Biohaven, BioResearch, BrainCells, Bristol-Myers Squibb, CeNeRx BioPharma, Cephalon, Cerecor, Clarus Funds, Clintara, Covance, Covidien, Eli Lilly, EnVivo, Euthymics Bioscience, Forest, FORUM, Ganeden Biotech, GlaxoSmithKline, Harvard Clinical Research Institute, Hoffman-LaRoche, Icon Clinical Research, i3 Innovus/Ingenix, Janssen, Jed Foundation, Johnson & Johnson, Lichtwer Pharma, Lorex, Lundbeck, Marinus, MedAvante, Methylation Sciences, National Alliance for Research on Schizophrenia and Depression, National Center for Complementary and Alternative Medicine, National Coordinating Center for Integrated Medicine, National Institute of Drug Abuse (NIDA), NIMH, Neuralstem, NeuroRx, Novartis, Organon, Otsuka, Pamlab, Pfizer; Pharmacia-Upjohn, Pharmaceutical Research Associates; Pharmavite, PharmoRx Therapeutics, Photothera, Reckitt Benckiser, Roche Pharmaceuticals, RCT Logic (formerly Clinical Trials Solutions), Sanofi-Aventis US, Shire, Solvay, Stanley Medical Research Institute, Synthelabo, Taisho, Takeda, Tal Medical, VistaGen, and Wyeth-Ayerst; has received advisory board/consultant fees from Abbott, ACADIA, Affectis, Alkermes, Amarin, Aspect Medical Systems, AstraZeneca, Auspex, Avanir, Axsome Therapeutics, Bayer, Best Practice Project Management, Biogen, BioMarin, Biovail, Boehringer Ingelheim, Boston Pharmaceuticals, BrainCells, Bristol-Myers Squibb, CeNeRx BioPharma, Cephalon, Cerecor, CNS Response, Compellis, Cypress, DiagnoSearch Life Sciences (P), Dainippon Sumitomo, Dov, Edgemont, Eisai, Eli Lilly, EnVivo, ePharmaSolutions, EPIX, Euthymics Bioscience, Fabre-Kramer, Forest, FORUM, GenOmind, GlaxoSmithKline, Grunenthal, Indivior, i3 Innovus/Ingenis, Intracellular, Janssen, Jazz, Johnson & Johnson, Knoll, Labopharm, Lorex, Lundbeck, Marinus, MedAvante, Merck, MSI Methylation Sciences, Naurex, Navitor, Nestle Health Sciences, Neuralstem, Neuronetics, NextWave, Novartis, Nutrition 21, Orexigen, Organon, Osmotica, Otsuka, Pamlab, Pfizer, PharmaStar, Pharmavite, PharmoRx Therapeutics, Praxis Precision Medicines, Precision Human Biolaboratory, Prexa, PPD, Purdue Pharma, Puretech Ventures, PsychoGenics, Psylin Neurosciences, RCT Logic (formerly Clinical Trials Solutions), Relmada Therapeutics, Rexahn, Ridge Diagnostics, Roche, Sanofi-Aventis US, Sepracor, Servier, Schering-Plough, Shenox, Solvay, Somaxon, Somerset, Sunovion, Supernus, Synthelabo, Taisho, Takeda, Tal Medical, Tetragenex, Teva, TransForm, Transcept, Usona Institute, Vanda, Versant Venture Management, and VistaGen; has received speaking/publishing fees from Adamed, Advanced Meeting Partners, American Psychiatric Association, American Society of Clinical Psychopharmacology, AstraZeneca, Belvoir Media Group, Boehringer Ingelheim, Bristol-Myers Squibb, Cephalon, CME Institute/Physicians Postgraduate Press, Eli Lilly, Forest Pharmaceuticals, GlaxoSmithKline, Imedex, MGH Psychiatry Academy/Primedia, MGH Psychiatry Academy/Reed Elsevier, Novartis, Organon, Pfizer, PharmaStar, United BioSource, and Wyeth-Ayerst; has equity holdings in Compellis and PsyBrain; receives royalty/patent, other income for patents for Sequential Parallel Comparison Design (SPCD), licensed by MGH to Pharmaceutical Product Development, LLC (PPD) (US_7840419, US_7647235, US_7983936, US_8145504, US_8145505); has a patent application for a combination of ketamine plus scopolamine in major depressive disorder (MDD), licensed by MGH to Biohaven; has patents for pharmacogenomics of depression treatment with folate (US_9546401, US_9540691); holds the copyright for the MGH Cognitive & Physical Functioning Questionnaire (CPFQ), Sexual Functioning Inventory (SFI), Antidepressant Treatment Response Questionnaire (ATRQ), Discontinuation-Emergent Signs & Symptoms (DESS), Symptoms of Depression Questionnaire (SDQ), and SAFER; and has received royalties from Lippincott, Williams & Wilkins; Wolters Kluwer; and World Scientific Publishing Co. Pte. Ltd; his disclosures (lifetime) can be viewed online at: http://mghcme.org/faculty/faculty-detail/maurizio_fava (updated September 2018). Dr Papakostas has received consulting fees from Abbott, Acadia*, Alkermes, AstraZeneca, Avanir, Axsome Therapeutics*, Brainsway, Bristol-Myers Squibb, Cephalon, Dey, Eli Lilly, Genentech*, Genomind*, GlaxoSmithKline, Evotec, H. Lundbeck, Inflabloc, Janssen*, Jazz, Johnson & Johnson*, Methylation Sciences, Mylan*, Novartis Pharma, One Carbon Therapeutics*, Osmotica*, Otsuka, Pamlab, Pfizer, Pierre Fabre, Ridge Diagnostics (formerly known as Precision Human Biolaboratories), Shire, Sunovion, Taisho, Takeda, Theracos, and Wyeth; has received honoraria (for lectures or consultancy) from Abbott, ACADIA, Alkermes, Asopharma Centroamerica y Caribe, AstraZeneca, Avanir, Bristol-Myers Squibb, Brainsway, Cephalon, Dey, Eli Lilly, Evotec, Forest, GlaxoSmithKline, Inflabloc, Grunbiotics, Jazz, H. Lundbeck, Medichem, Meiji Seika, Novartis, Otsuka, Pamlab, Pfizer, Pharma Trade, Pierre Fabre, Ridge Diagnostics, Shire, Sunovion, Takeda, Theracos, Titan, and Wyeth; and has received research support (paid to hospital) from AstraZeneca, Bristol-Myers Squibb, Forest, NIMH, Neuralstem*, Pamlab, Pfizer, Ridge Diagnostics (formerly known as Precision Human Biolaboratories), Sunovion, Tal Medical, and Theracos. He served (not currently) on the speakers bureau for Bristol-Myers Squibb and Pfizer. (*Asterisk denotes activity undertaken on behalf of Massachusetts General Hospital.) Dr Shelton has received grants from National Institutes of Health (NIH), Patient-Centered Outcomes Research Institute, ACADIA, Alkermes, Allergan, Avanir, Cerecor, Genomind, Intracellular Therapies, Janssen, Myriad Genetics, Navitor, NeuroRx, Novartis, Otsuka, Nestle’ Health, Novartis, and Takeda and has received consulting fees from ACADIA, Allergan, Cerecor, Clintara, Janssen, Lundbeck, Medtronic, MSI Methylation Sciences, Naurex, Nestle Health, Pfizer, and Takeda. In the past 3 years, Dr Thase has received advisory/consultant fees from ACADIA, Akili, Alkermes, Allergan (Forest, Naurex), Cerecor, Fabre-Kramer, Gerson Lehrman Group, Guidepoint Global, Johnson & Johnson (Janssen, Ortho-McNeil), H. Lundbeck, Moksha8, Nestlé (Pamlab), Neuralstem, Novartis, Otsuka, Pfizer, Sage, and Sunovion; has received grant support from ACADIA, Agency for Healthcare Research and Quality, Alkermes, Allergan (Forest, Naurex), AssureRx Health, Avanir, Axsome, Intracellular Therapies, Janssen, Johnson & Johnson (Janssen, Ortho-McNeil), NIH/NIMH, Otsuka, Patient-Centered Outcomes Research Institute, and Takeda; and has received royalties from American Psychiatric Foundation, Guilford Publications, Herald House, and W.W. Norton & Company. His spouse is employed by Peloton Advantage, which does business with most major pharmaceutical companies. Dr Trivedi has received advisor/consultant and received fees from Alkermes, AstraZeneca, Cerecor, Eli Lilly, Lundbeck, Naurex, Neuronetics, Otsuka, Pamlab, Pfizer Inc, Shire, and Takeda. and has received grants/research support from NIMH and NIDA. Drs Dirks, Liu, and Stankovic are employees of ACADIA Pharmaceuticals Inc, San Diego, California.
Funding/support: This study was supported by ACADIA Pharmaceuticals Inc, San Diego, California.
Role of the sponsor: The funder of the study had a role in study design, data analysis, data interpretation, and writing of the report.
Disclaimer: Dr Freeman, JCP Editor in Chief, was not involved in the editorial review or decision to publish this article.
Previous presentation: Presented at the 2019 American Psychiatric Association Annual Meeting, May 18-22, 2019, San Francisco, California.
Acknowledgments: The authors acknowledge the editorial assistance of Richard S. Perry, PharmD, in the preparation of this manuscript, which was supported by ACADIA Pharmaceuticals Inc, San Diego, California.
Supplementary material: See accompanying pages.
REFERENCES
1.Depression Fact Sheet. WHO website. https://www.who.int/news-room/fact-sheets/detail/depression. March 22, 2018. Accessed December 15, 2018.
2.Baldessarini RJ, Forte A, Selle V, et al. Morbidity in depressive disorders. Psychother Psychosom. 2017;86(2):65-72. PubMed CrossRef
3.Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 2018;391(10128):1357-1366. PubMed CrossRef
4.Machado-Vieira R, Henter ID, Zarate CA Jr. New targets for rapid antidepressant action. Prog Neurobiol. 2017;152:21-37. PubMed CrossRef
5.Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am. 1996;19(2):179-200. PubMed CrossRef
6.Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry. 2006;163(7):1161-1172. PubMed CrossRef
7.Papakostas GI. Managing partial response or nonresponse: switching, augmentation, and combination strategies for major depressive disorder. J Clin Psychiatry. 2009;70(suppl 6):16-25. PubMed CrossRef
8.Otte C, Gold SM, Penninx BW, et al. Major depressive disorder. Nat Rev Dis Primers. 2016;2(1):16065. PubMed CrossRef
9.Nelson JC, Papakostas GI. Atypical antipsychotic augmentation in major depressive disorder: a meta-analysis of placebo-controlled randomized trials. Am J Psychiatry. 2009;166(9):980-991. PubMed CrossRef
10.Mulder R, Hamilton A, Irwin L, et al. Treating depression with adjunctive antipsychotics. Bipolar Disord. 2018;20(suppl 2):17-24. PubMed CrossRef
11.Spielmans GI, Berman MI, Linardatos E, et al. Adjunctive atypical antipsychotic treatment for major depressive disorder: a meta-analysis of depression, quality of life, and safety outcomes. PLoS Med. 2013;10(3):e1001403. PubMed CrossRef
12.Zhou X, Keitner GI, Qin B, et al. Atypical antipsychotic augmentation for treatment-resistant depression: a systematic review and network meta-analysis. Int J Neuropsychopharmacol. 2015;18(11):pyv060. PubMed CrossRef
13.Fava M, Memisoglu A, Thase ME, et al. Opioid modulation with buprenorphine/samidorphan as adjunctive treatment for inadequate response to antidepressants: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2016;173(5):499-508. PubMed CrossRef
14.Vanover KE, Weiner DM, Makhay M, et al. Pharmacological and behavioral profile of N-(4-fluorophenylmethyl)-N-(1-methylpiperidin-4-yl)-N‘ ²-(4-(2-methylpropyloxy)phenylmethyl) carbamide (2R,3R)-dihydroxybutanedioate (2:1) (ACP-103), a novel 5-hydroxytryptamine(2A) receptor inverse agonist. J Pharmacol Exp Ther. 2006;317(2):910-918. PubMed CrossRef
15.Artigas F. Developments in the field of antidepressants, where do we go now? Eur Neuropsychopharmacol. 2015;25(5):657-670. PubMed CrossRef
16.Artigas F, Bortolozzi A, Celada P. Can we increase speed and efficacy of antidepressant treatments? part I: general aspects and monoamine-based strategies. Eur Neuropsychopharmacol. 2018;28(4):445-456. PubMed CrossRef
17.Marek GJ, Carpenter LL, McDougle CJ, et al. Synergistic action of 5-HT2A antagonists and selective serotonin reuptake inhibitors in neuropsychiatric disorders. Neuropsychopharmacology. 2003;28(2):402-412. PubMed CrossRef
18.Marek GJ, Li AA, Seiden LS. Evidence for involvement of 5-hydroxytryptamine1 receptors in antidepressant-like drug effects on differential-reinforcement-of-low-rate 72-second behavior. J Pharmacol Exp Ther. 1989;250(1):60-71. PubMed
19.Desseilles M, Witte J, Chang TE, et al. Massachusetts General Hospital SAFER criteria for clinical trials and research. Harv Rev Psychiatry. 2013;21(5):269-274. PubMed CrossRef
20.Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382-389. PubMed CrossRef
21.Guy W. Clinical Global Impressions. ECDEU Assessment Manual for Psychopharmacology, Revised (DHEW Publication No. ADM 76-338). Rockville, MD: US Department of Health, Education, and Welfare; Public Health Service; Alcohol, Drug Abuse, and Mental Health Administration; 1976:217-222.
22.Chandler GM, Iosifescu DV, Pollack MH, et al. RESEARCH: Validation of the Massachusetts General Hospital Antidepressant Treatment History Questionnaire (ATRQ). CNS Neurosci Ther. 2010;16(5):322-325. PubMed CrossRef
23.Fava M, Evins AE, Dorer DJ, et al. The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach. Psychother Psychosom. 2003;72(3):115-127. PubMed CrossRef
24.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56-62. PubMed CrossRef
25.First MB, Williams JBW, Karg RS, et al. Structured Clinical Interview for DSM-5 Disorders-Clinicial Trials Version (SCID-5-CT). Arlington, VA: American Psychiatric Association; 2015.
26.Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996;11(suppl 3):89-95. PubMed CrossRef
27.Kaida K, Takahashi M, Akerstedt T, et al. Validation of the Karolinska sleepiness scale against performance and EEG variables. Clin Neurophysiol. 2006;117(7):1574-1581. PubMed CrossRef
28.Ware J Jr, Kosinski M, Keller SDA. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care. 1996;34(3):220-233. PubMed CrossRef
29.Hogan TP, Awad AG, Eastwood R. A self-report scale predictive of drug compliance in schizophrenics: reliability and discriminative validity. Psychol Med. 1983;13(1):177-183. PubMed CrossRef
30.Labbate LA, Lare SB. Sexual dysfunction in male psychiatric outpatients: validity of the Massachusetts General Hospital Sexual Functioning Questionnaire. Psychother Psychosom. 2001;70(4):221-225. PubMed CrossRef
31.Patton JH, Stanford MS, Barratt ES. Factor structure of the Barratt impulsiveness scale. J Clin Psychol. 1995;51(6):768-774. PubMed CrossRef
32.Mannix S, Hassan M, Tummala R, et al. Content validity of the Sheehan Irritability Scale in patients with major depressive disorder. Int Clin Psychopharmacol. 2016;31(2):110-117. PubMed CrossRef
33.Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266-1277. PubMed CrossRef
34.Barnes TR. The Barnes Akathisia Rating Scale—revisited. J Psychopharmacol. 2003;17(4):365-370. PubMed CrossRef
35.Guy W. Abnormal Involuntary Movement Scale. ECDEU Assessment Manual for Psychopharmacology, Revised (DHEW Publication No. ADM 76-338). Rockville, MD: US Department of Health, Education, and Welfare; Public Health Service; Alcohol, Drug Abuse, and Mental Health Administration; 1976:534-537.
36.Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand suppl. 1970;212:11-19. PubMed CrossRef
37.Khan A, Fahl Mar K, Faucett J, et al. Has the rising placebo response impacted antidepressant clinical trial outcome? data from the US Food and Drug Administration 1987-2013. World Psychiatry. 2017;16(2):181-192. PubMed CrossRef
38.Furukawa TA, Cipriani A, Atkinson LZ, et al. Placebo response rates in antidepressant trials: a systematic review of published and unpublished double-blind randomised controlled studies. Lancet Psychiatry. 2016;3(11):1059-1066. PubMed CrossRef
39.Papakostas GI, טstergaard SD, Iovieno N. The nature of placebo response in clinical studies of major depressive disorder. J Clin Psychiatry. 2015;76(4):456-466. PubMed CrossRef
40.Trivedi MH, South C, Jha MK, et al. A novel strategy to identify placebo responders: prediction index of clinical and biological markers in the EMBARC Trial. Psychother Psychosom. 2018;87(5):285-295. PubMed CrossRef
41.Lee Y, Rosenblat JD, Lee J, et al. Efficacy of antidepressants on measures of workplace functioning in major depressive disorder: a systematic review. J Affect Disord. 2018;227:406-415. PubMed CrossRef
42.Weiller E, Weiss C, Watling CP, et al. Functioning outcomes with adjunctive treatments for major depressive disorder: a systematic review of randomized placebo-controlled studies. Neuropsychiatr Dis Treat. 2017;14:103-115. PubMed CrossRef
43.Sheehan DV, Nakagome K, Asami Y, et al. Restoring function in major depressive disorder: a systematic review. J Affect Disord. 2017;215:299-313. PubMed CrossRef
44.Ballard C, Banister C, Khan Z, et al; ADP Investigators. Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double-blind study. Lancet Neurol. 2018;17(3):213-222. PubMed CrossRef
45.Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet. 2014;383(9916):533-540. PubMed CrossRef
46.Mohamed S, Johnson GR, Chen P, et al; VAST-D Investigators. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: The VAST-D Randomized Clinical Trial. JAMA. 2017;318(2):132-145. PubMed CrossRef
47.Solmi M, Murru A, Pacchiarotti I, et al. Safety, tolerability, and risks associated with first- and second-generation antipsychotics: a state-of-the-art clinical review. Ther Clin Risk Manag. 2017;13:757-777. PubMed CrossRef
48.Yoon S, Jeon SW, Ko YH, et al. Adjunctive brexpiprazole as a novel effective strategy for treating major depressive disorder: a systematic review and meta-analysis. J Clin Psychopharmacol. 2017;37(1):46-53. PubMed CrossRef
49.Clayton AH, El Haddad S, Iluonakhamhe J-P, et al. Sexual dysfunction associated with major depressive disorder and antidepressant treatment. Expert Opin Drug Saf. 2014;13(10):1361-1374. PubMed CrossRef
50.Henssler J, Kurschus M, Franklin J, et al. Trajectories of acute antidepressant efficacy: how long to wait for response? a systematic review and meta-analysis of long-term, placebo-controlled acute treatment trials. J Clin Psychiatry. 2018;79(3):17r11470. PubMed CrossRef
This PDF is free for all visitors!




