Objective: To investigate the efficacy and safety of levomilnacipran sustained release (SR), an antidepressant candidate in late-stage development, in major depressive disorder (MDD).
Method: Between December 2006 and October 2007, a 10-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter, flexible-dose trial assessed once-daily levomilnacipran SR (75 mg or 100 mg) in outpatients (18-70 years) meeting DSM-IV criteria for a major depressive episode (duration ≥ 1 month) with a 17-item Hamilton Depression Rating Scale (HDRS17) score > 22 and Sheehan Disability Scale (SDS) score ≥ 10. Levomilnacipran SR dose was increased to 100 mg/d over 12 days. The primary efficacy measure was Montgomery Asberg Depression Rating Scale (MADRS) score change from baseline to week 10; secondary efficacy measures were the HDRS17, SDS, Clinical Global Impressions-Improvement scale, and MADRS response (≥ 50% decrease from baseline) and remission (score ≤ 10). Safety was evaluated according to adverse events, laboratory investigations, and vital signs/physical findings.
Results: Efficacy analyses included 276 levomilnacipran SR-treated patients and 277 placebo-treated patients. Levomilnacipran SR was significantly superior to placebo on MADRS total score change from baseline to week 10 (least squares mean difference [LSMD] = −4.2 [95% CI, −5.7 to −2.6]; P < .0001). Statistical significance in favor of levomilnacipran SR was demonstrated on change from baseline to week 10 in HDRS17 total score (LSMD = −3.4 [95% CI, −4.7 to −2.2]; P < .0001) and SDS total score (LSMD = −3.4 [95% CI, −4.6 to −2.2]; P < .0001) and subscales. Significantly more levomilnacipran SR patients versus placebo patients achieved MADRS response (59.1% vs 42.2%; P < .0001) and remission (46.4% vs 26.0%; P < .0001). Levomilnacipran SR was generally safe and well tolerated; more levomilnacipran SR patients (9.4%) versus placebo patients (6.5%) discontinued due to adverse events, but more placebo patients versus levomilnacipran SR patients discontinued overall (24.9% vs 20.2%).
Conclusions: Levomilnacipran SR demonstrated robust efficacy on all measures and was generally well tolerated.
Trial Registration: EudraCT number: 2006-002404-34
J Clin Psychiatry 2013;74(4):363-369
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: August 29, 2012; accepted February 1, 2013 (doi:10.4088/JCP.12m08141).
Corresponding author: Stuart A. Montgomery, MD, PO Box 8751, London W13 8WH, UK ([email protected]).
Efficacy and Safety of Levomilnacipran Sustained Release in Moderate to Severe Major Depressive Disorder: A Randomized, Double-Blind, Placebo-Controlled, Proof-of-Concept Study
ABSTRACT
Objective: To investigate the efficacy and safety of levomilnacipran sustained release (SR), an antidepressant candidate in late-stage development,
in major depressive disorder (MDD).
Method: Between December 2006 and October 2007, a 10-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter, flexible-dose trial assessed once-daily levomilnacipran SR (75 mg or 100 mg) in outpatients (18–70 years) meeting DSM-IV criteria for a major depressive episode (duration ≥ 1 month) with a 17-item Hamilton Depression Rating Scale (HDRS17) score > 22 and Sheehan Disability Scale (SDS) score ≥ 10. Levomilnacipran SR dose was increased to 100 mg/d over 12 days. The primary efficacy measure was Montgomery Asberg Depression Rating Scale (MADRS) score change from baseline to week 10; secondary efficacy measures were the HDRS17, SDS, Clinical Global Impressions-Improvement scale, and MADRS response (≥ 50% decrease from baseline) and remission (score ≤ 10). Safety was evaluated according to adverse events, laboratory investigations, and vital signs/physical findings.
Results: Efficacy analyses included 276 levomilnacipran SR–treated patients and 277 placebo-treated patients. Levomilnacipran SR was significantly superior to placebo on MADRS total score change from baseline to week 10 (least squares mean difference [LSMD] = −4.2 [95% CI, −5.7 to −2.6]; P < .0001). Statistical significance in favor of levomilnacipran SR was demonstrated on change from baseline to week 10 in HDRS17 total score (LSMD = −3.4 [95% CI, −4.7 to −2.2]; P < .0001) and SDS total score (LSMD = −3.4 [95% CI, −4.6 to −2.2]; P < .0001) and subscales. Significantly more levomilnacipran SR patients versus placebo patients achieved MADRS response (59.1% vs 42.2%; P < .0001) and remission (46.4% vs 26.0%; P < .0001). Levomilnacipran SR was generally safe and well tolerated; more levomilnacipran SR patients (9.4%) versus placebo patients (6.5%) discontinued due to adverse events, but more placebo patients versus levomilnacipran SR patients discontinued overall (24.9% vs 20.2%).
Conclusions: Levomilnacipran SR demonstrated robust efficacy on all measures and was generally
well tolerated.
Trial Registration: EudraCT number: 2006-002404-34
J Clin Psychiatry 2013;74(4):363–369
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: August 29, 2012; accepted February 1, 2013 (doi:10.4088/JCP.12m08141).
Corresponding author: Stuart A. Montgomery, MD, PO Box 8751, London W13 8WH, UK ([email protected]).
Major depressive disorder (MDD) is a common, potentially dangerous, and disabling disorder. It has a chronic recurrent course and is recognized as the most common cause of disability during the course of a working life.1 Effective treatment of MDD is a public health priority.
Levomilnacipran (1S, 2R-milnacipran), a potent and selective
serotonin-norepinephrine reuptake inhibitor (SNRI) with greater potency for inhibition of norepinephrine (NE) relative to serotonin (5-HT) reuptake, is in clinical development for the treatment of adult MDD. In animal models of depression, it shows a potent antidepressant-like effect2 suggesting that it may offer therapeutic effects similar to older tricyclic antidepressants without the accompanying safety and tolerability issues. The sustained release (SR) formulation was developed to allow for once-daily dosing.
Potency for inhibition of 5-HT reuptake relative to NE reuptake varies among members of the SNRI class of antidepressants. For example, 5-HT reuptake inhibition is 10-fold more potent than NE reuptake inhibition for duloxetine and 30-fold more potent for venlafaxine.3 Because NE reuptake inhibition for venlafaxine at lower doses is trivial, with meaningful NE reuptake inhibition achieved only at high doses,2 venlafaxine in effect becomes a selective serotonin reuptake inhibitor (SSRI) when prescribed at the usual dose of 150 mg/d or less. These relative differences make generalizing from one SNRI to another potentially misleading.
In contrast, levomilnacipran has approximately 2-fold higher potency for NE relative to 5-HT reuptake inhibition and over 10-fold higher selectivity for NE reuptake inhibition compared with duloxetine and venlafaxine.2 While the general question of whether SNRIs have superior efficacy relative to SSRIs, particularly on the core symptoms of
depression, remains unresolved, the potentially greater efficacy of
SNRIs with greater inhibition of NE versus 5-HT reuptake is a more
specific area of investigation that is of particular interest in regard to
levomilnacipran SR.
The objective of this study (EudraCT number: 2006-002404-34) was to investigate the efficacy and safety of a flexible-dose regimen of levomilnacipran SR compared with placebo in the treatment of MDD over a 10-week period.
METHOD
Patients were recruited to participate in the study from 68 sites in France, Finland, Latvia, Lithuania, Sweden, Germany, Estonia, Czech Republic, Bulgaria, India, and South Africa; the study was conducted between December 13, 2006, and October 22, 2007. The final study protocol was approved by ethics committees and appropriate authorities for all centers involved. The study was performed in accordance with the principles stated in the Declaration of Helsinki and Good Clinical Practice guidelines, as applicable at the time; all patients provided
written, informed consent.
Study Design
This 10-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter, flexible-dose trial assessed once-daily levomilnacipran SR (75 mg or 100 mg) in adult patients with MDD. Patients were randomized by a computer-generated list of numbers that were blindly linked to test drug or placebo; groups in each study center were balanced according to severity of baseline depression (Montgomery Asberg Depression Rating Scale [MADRS]4 baseline total scores < 30 or ≥ 30). A 3- to 21-day drug washout period was followed by 2-week progressive titration, an 8-week double-blind treatment period, and a 1-week down-titration period.
During titration, patients received an equivalent number of identical-looking levomilnacipran SR or placebo capsules. Patients randomly assigned to levomilnacipran SR received 25 mg on days 1–3, 50 mg on days 4–7, and 75 mg on days 8–11. If good tolerance was discerned by telephone assessment, the 100-mg levomilnacipran SR target dose was initiated on day 12 and maintained for the study duration. If subsequent intolerance developed, dose reduction to 75 mg was allowed and the dose was fixed for the remainder of the trial; patients were withdrawn from the study if the 75-mg dose was not tolerated. Throughout comparative treatment, capsules that corresponded to the randomized treatment number were dispensed; blinded information was maintained in sealed decoding envelopes. At the end of double-blind treatment or at premature withdrawal, patients were down-titrated for 7 days (50 mg for 4 days; 25 mg for 3 days) and followed up for an additional 7 days.
Patients
Inclusion and exclusion criteria. Male or female outpatients 18–70 years of age who met criteria for an episode of MDD (moderate or severe, without psychotic features) as defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR)5 were eligible for study participation. The duration of the current episode was required to be ≥ 1 month; MDD was confirmed using the Mini-International Neuropsychiatric Interview6; inclusion required a 17-item Hamilton Depression Rating Scale (HDRS17)7 score > 22 and a Sheehan Disability Scale (SDS)8 score ≥ 10, with at least 1 subscale (work, social life, or family life) score ≥ 6. Women of childbearing potential were required to use a medically accepted method of contraception.
No clinically relevant abnormalities in clinical examination, laboratory test, or electrocardiography (ECG) findings were allowed. Additional reasons for exclusion included history of psychotic or bipolar disorder, various other current psychiatric (including generalized anxiety disorder and social phobia preceding the onset of the depressive episode) or personality disorders (categorized according to DSM-IV clusters), substance abuse (preceding 6 months) or dependence (preceding 2 years), physical conditions (eg, cardiovascular disease, systemic disease, history of seizure disorder), pregnancy, allergy/nonresponse to milnacipran, initiating or stopping formal psychotherapy (preceding 6 months), electroconvulsive therapy (preceding 3 months), moderate/severe suicide risk, and nonresponse to 2 previous antidepressants of adequate dose for ≥ 4 weeks for the current depressive episode. Concomitant psychotropic medications were prohibited except for limited use of hypnotics (ie, zolpidem, zopiclone), low-dose neuroleptics, or low-dose anxiolytics (10-mg diazepam equivalent) under specific circumstances (eg, if chronic treatment was initiated 3 months before the study, the medication regimen was maintained).
Efficacy Assessments
Patients were assessed with the MADRS4 at selection (day −21 to day −3), at inclusion (week 0), at all study weeks (weeks 1–4, 6, 8, 10), and after down-titration (weeks 11, 12) or premature withdrawal. The HDRS17 (weeks 0–4, 6, 8, 10–12), Clinical Global Impressions-Improvement (CGI-I) (weeks 2, 4, 6, 8, 10–12) and -Severity of Illness (CGI-S)9 (weeks 0, 2, 4, 8, 10), SDS total score and subscales (weeks 0, 2, 4, 6, 8, 10), MADRS response (≥ 50% baseline total score decrease) and remission (total score ≤ 10), CGI-I response (CGI score of 1 or 2), and HDRS17 response (≥ 50% baseline total score decrease) and remission (total score ≤ 7) were also evaluated.
Safety Assessments
Treatment-emergent adverse events (TEAEs) were evaluated at each visit via spontaneous reporting, nonleading questions, and clinical evaluation; they were categorized by relationship to study drug and intensity. Laboratory investigations (weeks 0, 4), ECGs (weeks 0, 4, 10), and vital signs/physical examinations (all visits) were recorded. Potentially significant changes from baseline in safety measures were predefined by low and high limits.
Statistical Analyses
The safety data set, consisting of all randomized patients who received at least 1 dose of study drug, was used for all safety evaluations. The full analysis set (FAS), which comprised all patients in the safety data set with at least 1 postbaseline evaluation of MADRS total score, was used for efficacy analyses.
The primary efficacy parameter was MADRS total score change from baseline to week 10; analysis was conducted using a mixed-effects model for repeated measures (MMRM) on the FAS. The model included treatment, center, and visit as main effects; MADRS total score baseline as covariate; and treatment-by-visit and baseline-by-visit interactions. Sensitivity analyses were performed using the last-observation-carried-forward (LOCF) approach and an analysis of covariance (ANCOVA) model with treatment and center as main effects and baseline MADRS total score as covariate.
Secondary efficacy parameters included change from baseline to week 10 in HDRS17 total score and SDS total/subscale scores; analyses were conducted using an MMRM model and ANCOVA similar to the primary analysis. CGI-I score was analyzed using MMRM analysis, excluding baseline and interaction with baseline term from the model. The Covi Anxiety Scale10 was used to measure anxiety; the change from baseline was summarized using descriptive statistics. Response and remission at week 10 were analyzed by the Cochran-Mantel-Haenszel test stratified by center at the end of week 10. Post hoc evaluations included the number needed to treat (NNT) analyses for response and remission as determined by the reciprocal of the difference of the event proportions between the levomilnacipran SR group and the placebo group.11 Statistical significance was determined by 2-sided tests performed at the 5% level. Descriptive statistics were used to evaluate safety outcomes.
RESULTS
Patient Disposition and Demographic Characteristics
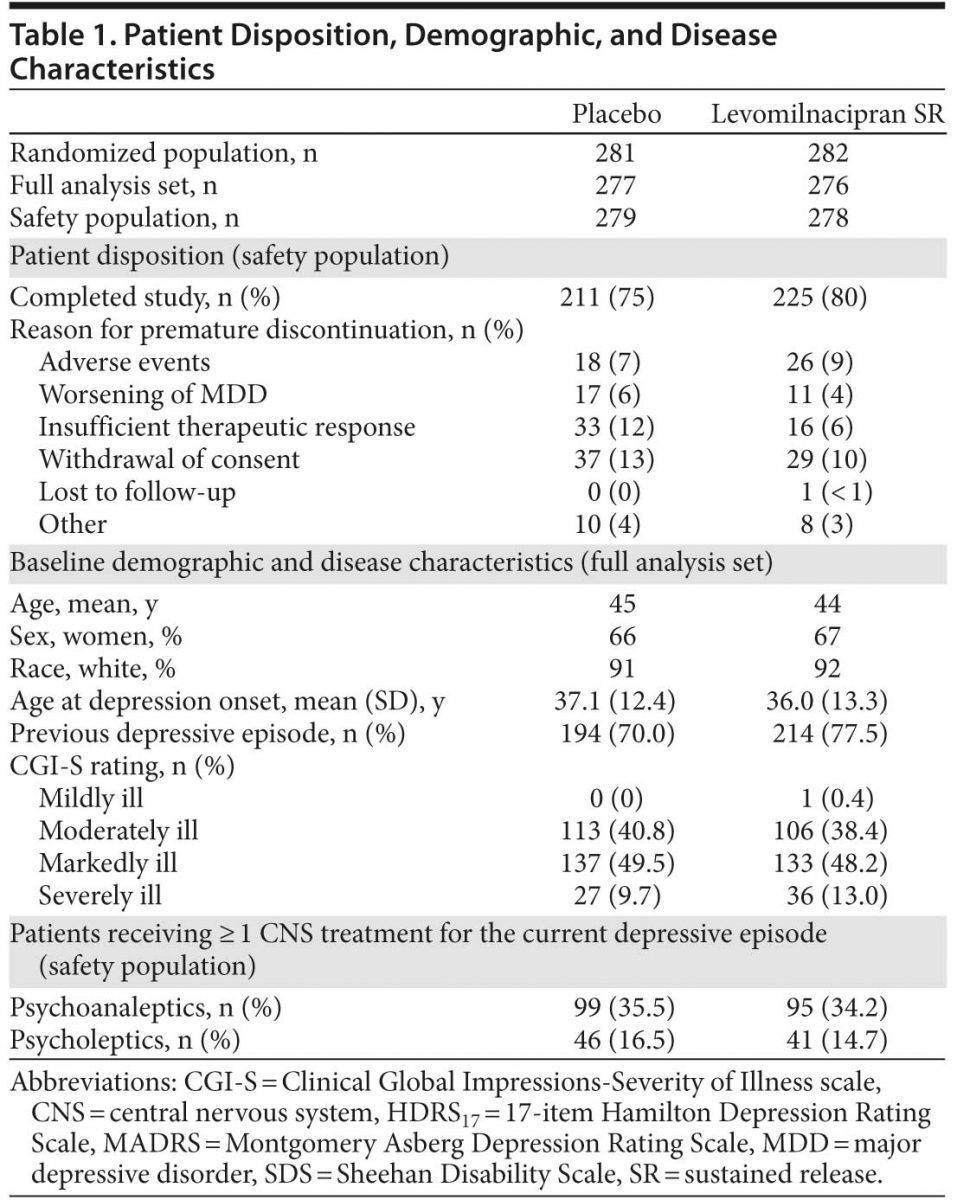
A total of 659 patients were screened for potential study participation; failure to meet at least 1 eligibility criterion was the most common reason for screen failure (56 patients). A total of 563 patients were randomized to treatment; 557 patients were in the safety data set, and 553 patients were in the FAS. A higher percentage of levomilnacipran SR patients completed the study; baseline demographic and disease characteristics and premature discontinuation for any reason were similar between treatment groups (Table 1). CGI-S baseline scores indicated that the majority of patients were moderately or markedly ill.
Following titration, 189 levomilnacipran SR patients (71.6%) were taking the 100-mg/d target dosage; during the study, 18 (9.5%) of these patients down-titrated to 75 mg/d. In the placebo group, 219 patients (81.4%) reached maximum dosage by the end of titration, and 10 (4.6%) down-titrated during the study. The mean (SD) exposure to treatment was 67.9 (20.7) days for placebo and 68.6 (20.9) days for levomilnacipran SR.
Efficacy
Primary analysis. Levomilnacipran SR was significantly superior to placebo on the primary efficacy parameter, MADRS total score change from baseline to week 10 (Table 2, Figure 1A). Mean MADRS score decreased throughout the study in both groups; a significantly greater decrease in favor of levomilnacipran SR was seen from week 3 onward.
Sensitivity analysis using the LOCF approach and an ANCOVA model in the FAS was supportive of the primary analysis (P < .0001); the magnitude of change was slightly smaller using LOCF analysis, but between-group differences were similar (least squares mean difference [LSMD] = −3.7 [−5.2 to −2.1]).
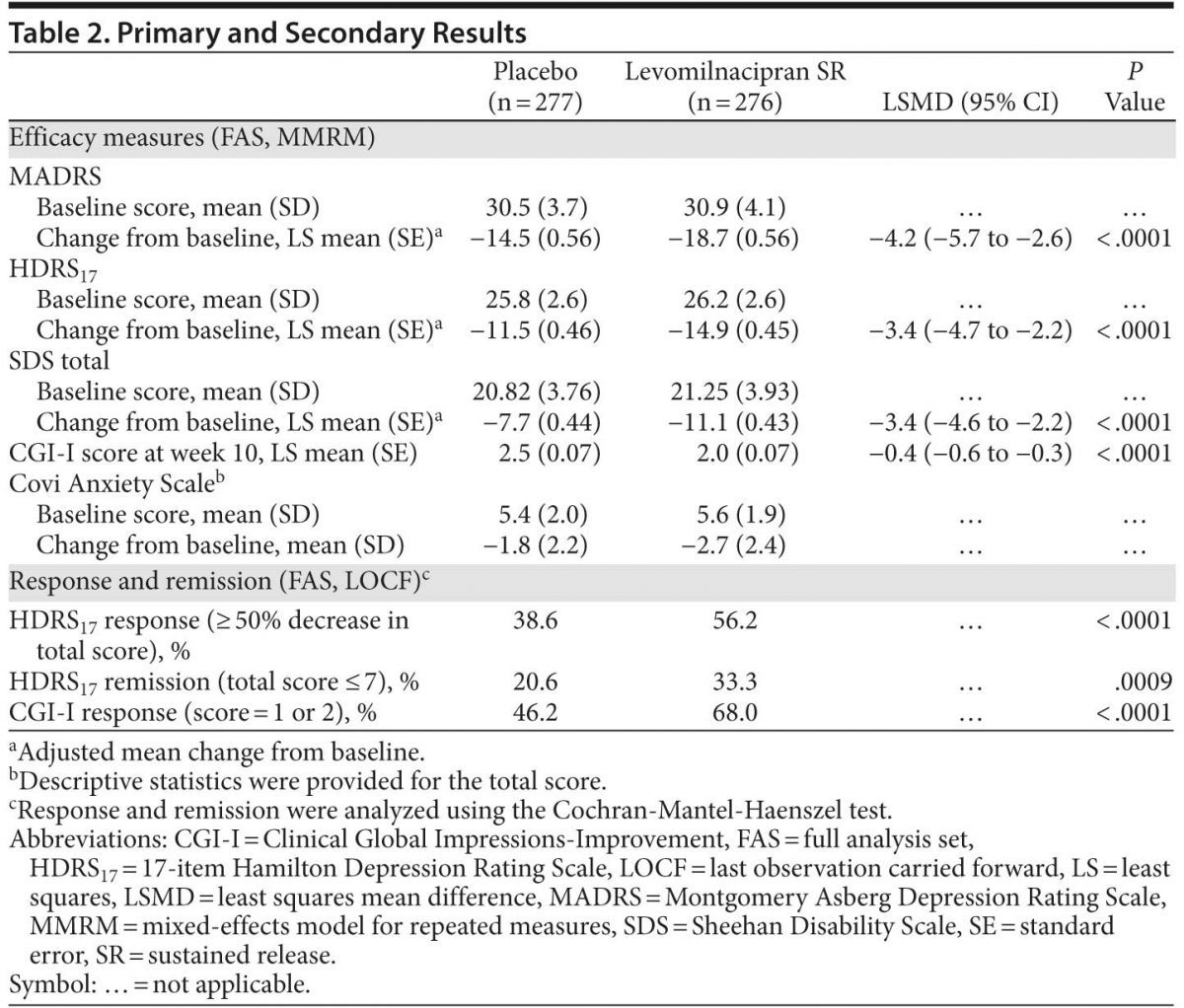
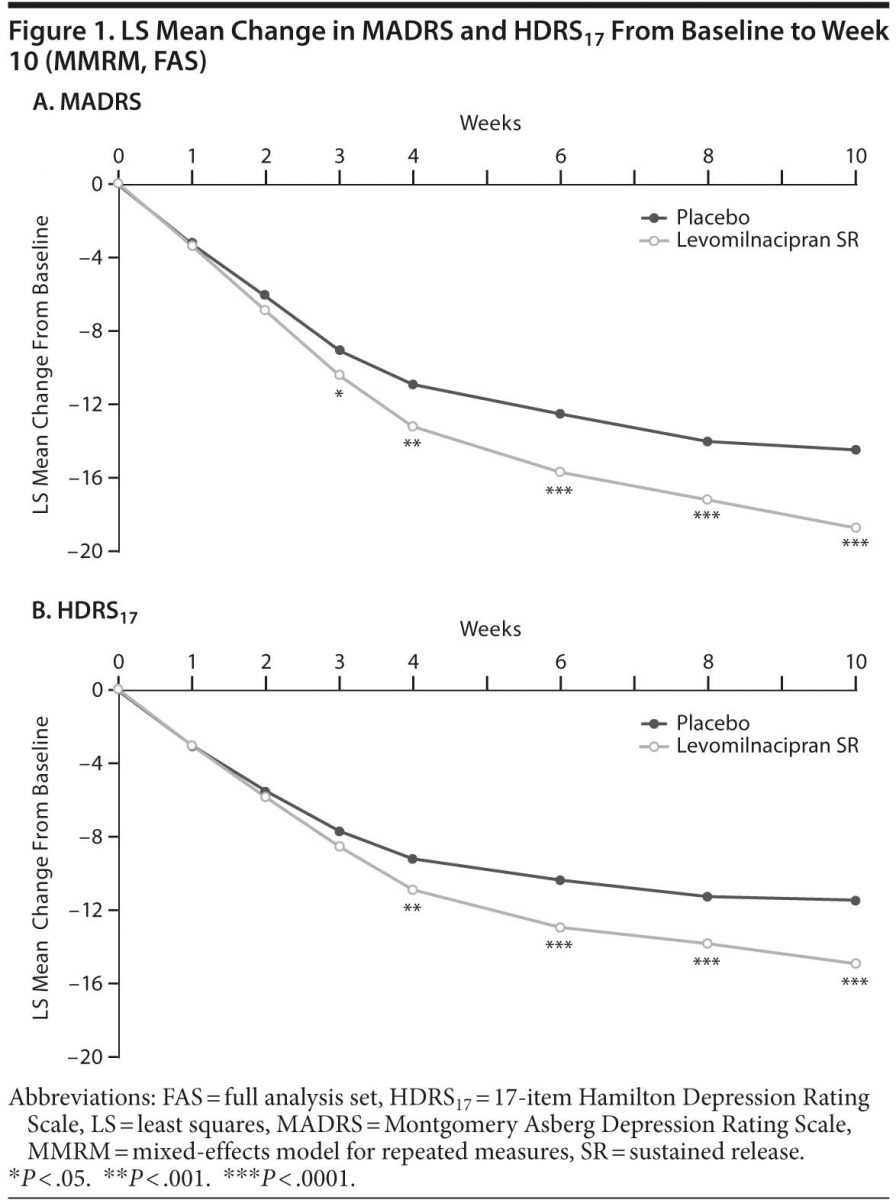
Secondary efficacy. Improvement on HDRS17 total score from baseline to week 10 was significantly superior for levomilnacipran SR versus placebo (Table 2, Figure 1B); sensitivity analysis using the LOCF approach supported MMRM results (LSMD = −2.8 [−4.1 to −1.6]; P < .0001).
Change from baseline to week 10 was significantly greater for levomilnacipran SR versus placebo on SDS total score (Table 2, Figure 2A); significant improvement in favor of levomilnacipran SR was also demonstrated on the work (LSMD = −1.1 [−1.5 to −0.7]; P < .0001), social life (LSMD = −1.0 [−1.5 to −0.6]; P < .0001), and family life (LSMD = −1.2 [−1.6 to −0.8]; P < .0001) subscales (Figure 2A). LOCF analyses supported the primary MMRM results on all SDS measures. Adjusted mean CGI-I score at week 10 was also significantly better for levomilnacipran SR than placebo. The mean change in Covi Anxiety Scale total score at week 10 was greater in the levomilnacipran SR group relative to placebo (Table 2).
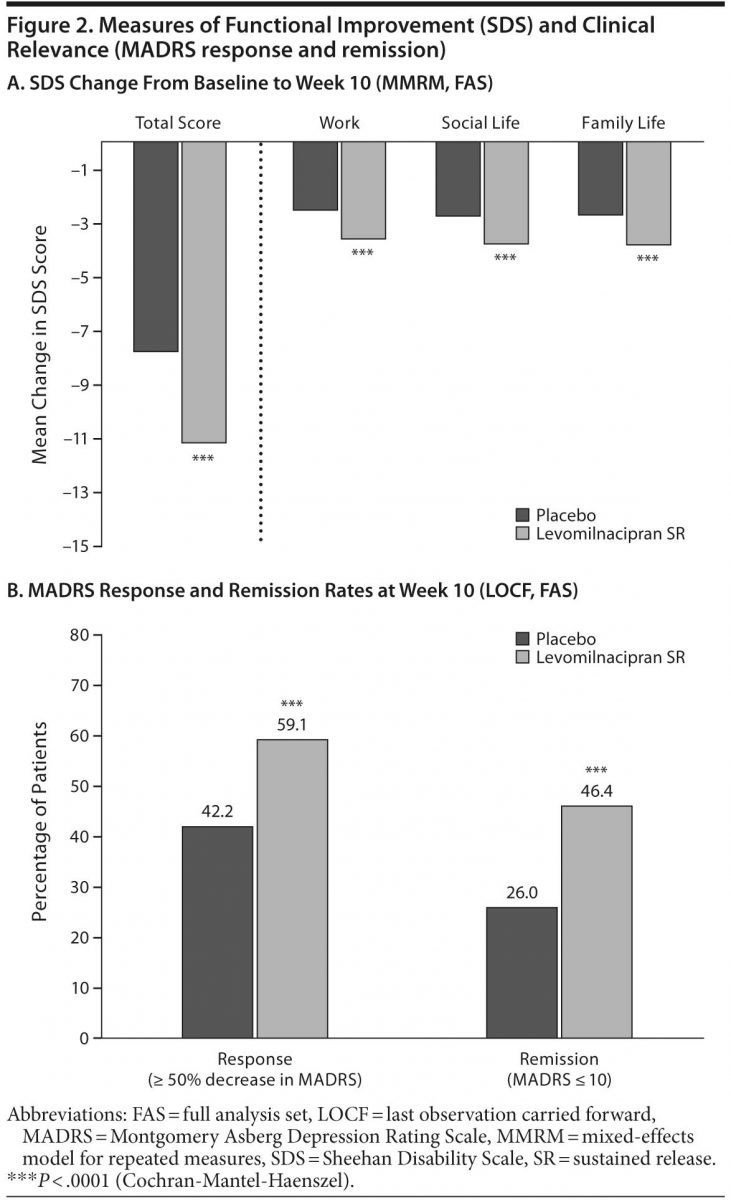
Response and remission. A significantly greater proportion of patients treated with levomilnacipran SR versus placebo attained MADRS response (≥ 50% decrease from baseline) and remission (total score ≤ 10) at week 10 (Figure 2B); there were significantly more levomilnacipran SR responders and remitters from week 6 onward. The NNTs (95% CI) for response and remission for levomilnacipran SR relative to placebo were 6 (4–12) and 5 (4–8), respectively.
The proportion of patients with HDRS17 response (≥ 50% decrease from baseline) was significantly greater for levomilnacipran SR compared with placebo; HDRS17 remission (total score ≤ 7) was also significantly greater for patients receiving levomilnacipran SR than those receiving placebo (Table 2), with more levomilnacipran SR patients remitting from week 3 onward. CGI-I response (score = 1 or 2) was significantly greater for levomilnacipran SR patients versus placebo patients (Table 2), with fewer responders in the placebo group at every study visit after week 2.
Safety
Adverse events. A summary of adverse events (AEs) and common TEAEs (≥ 5% in either group) is presented in Table 3. TEAEs that occurred in ≥ 5% of levomilnacipran SR patients with at least twice the frequency of placebo were hyperhidrosis, constipation, diarrhea, tachycardia, palpitations, and hypertension.
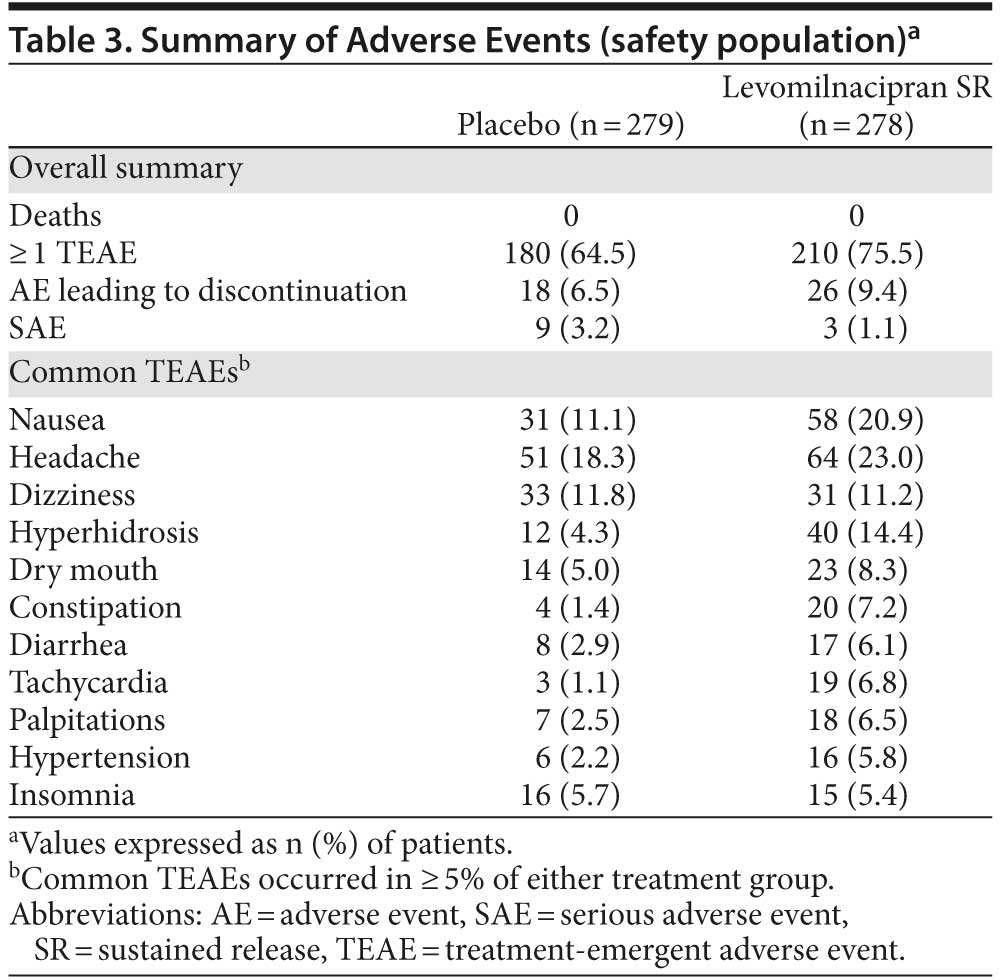
Nine serious AEs (SAEs) were reported by 9 patients in the placebo group (suicidal ideation, depression, and major depression [2 patients each]; depressive symptom, stress, and open fracture [1 patient each]); all placebo-related SAEs except for depressive symptom and open fracture resulted in discontinuation. Four SAEs were reported by 3 levomilnacipran SR patients (suicidal ideation [resulted in premature discontinuation], prostatitis and seminal vesiculitis [1 patient], and intervertebral disc protrusion).
Overall, 18 placebo patients (6.5%) and 26 levomilnacipran SR patients (9.4%) withdrew from the study due to AEs. The most common AE leading to discontinuation in the placebo group was suicidal ideation (4 patients). The most common AEs leading to discontinuation for levomilnacipran SR patients were nausea (5 patients) and vomiting (4 patients). During down-titration/treatment discontinuation, TEAE incidence was similar for placebo (8.2%) and levomilnacipran SR (8.6%), and no new SAEs were reported.
Clinical and laboratory evaluations. For levomilnacipran SR, there were no clinically relevant changes from baseline to week 10 in hematology or biochemistry values except
for slight mean (SD) increases in aspartate aminotransferase (placebo, 0.1 [6.8] U/L; levomilnacipran SR, 1.3 [8.0] U/L), alanine aminotransferase (placebo, 0.5 [10.1] U/L; levomilnacipran SR, 2.5 [12.2] U/L), and γ-glutamyl transferase (placebo, 0.6 [8.7] U/L; levomilnacipran SR, 2.1 [16.8] U/L). Mean changes in blood pressure from baseline to week 10 were similar between groups; mean increases in heart rate were noted for levomilnacipran SR (placebo, −1.2 [8.9] bpm; levomilnacipran SR, +7.0 [11.5] bpm).
Overall, clinically significant changes in vital signs were reported in 10 placebo patients and 18 levomilnacipran SR patients; changes in systolic and diastolic blood pressure and increases in heart rate were more common in levomilnacipran SR patients than placebo patients. The most common clinically significant changes for levomilnacipran SR were systolic blood pressure decrease (maximum −40 mm Hg; 6 patients each) and heart rate increase (maximum 42 bpm).
Based on the Fridericia correction (QTcF), there was no QTc prolongation (mean [SD] change from baseline to week 10/premature withdrawal: placebo, 0.4 [15.4] msec; levomilnacipran SR, –3.1 [15.4] msec). Based on the Bazett correction (QTcB), greater mean change from baseline was observed for levomilnacipran SR (8.0 [18.1] msec) versus placebo (−0.2 [19.1] msec), consistent with the increase in heart rate (placebo, −0.1 [9.2] bpm; levomilnacipran SR, 4.9 [9.8] bpm). There were no QTcF or QTcB increases ≥ 60 msec12 in the levomilnacipran SR group; QTcB and QTcF increases ≥ 60 msec were observed in zero and 1 placebo patient, respectively.
DISCUSSION
This 10-week, randomized, double-blind, placebo-controlled trial demonstrated robust treatment effect for levomilnacipran SR 75–100 mg/d compared with placebo in treating patients with MDD. A 4.2-point difference in favor of levomilnacipran SR versus placebo was demonstrated on MADRS total score change from baseline to week 10 (MMRM), the primary efficacy parameter. The treatment effect was considerably larger than the 2-point MADRS or HDRS drug-placebo difference commonly reported for antidepressants evaluated in meta-analyses of largely positive pivotal studies submitted to regulators in the United States and Europe13–15 and demonstrates that patients treated with levomilnacipran SR benefit from clinically relevant improvement.
Large and significant differences in favor of levomilnacipran SR were also observed on all secondary measures. Similar to MADRS outcomes, results on the HDRS17
demonstrated robust symptom improvement in patients treated with levomilnacipran SR. Additionally, significant improvement for levomilnacipran SR on the SDS and its subscales demonstrated efficacy in treating functional impairment, a by-product of depression that results in decreased psychosocial and work functioning.16 Even though functional improvement may lag behind symptom improvement, this outcome is an important component of wellness,17 and these results suggest that levomilnacipran SR plays an important role in a return to normal functioning.
Response and remission rates were significantly greater for levomilnacipran SR compared with placebo using several different measures. Response far exceeded the 10% average advantage for drug versus double-blind placebo reported in most registration trials of newer antidepressants.18 Remission, an important and clinically relevant outcome,19 occurred in 46% of levomilnacipran SR–treated patients and 26% of placebo-treated patients when remission was defined using MADRS criteria; HDRS17 remission results were in accord. The 20% absolute difference in remission for levomilnacipran SR compared with placebo is noteworthy because studies of this type are not normally powered for analysis of this outcome measure and remission is difficult to achieve in the timeframe of a short-term study.19
The NNT, the number of patients that would need to be treated to observe 1 additional successful outcome, is the effect size that best reflects clinical significance for binary outcomes like response and remission.20 For antidepressants, an NNT of ≤ 10 for response is considered the threshold at which a clinically relevant treatment advantage is indicated.19,21 In this study, the NNT based on the MADRS for levomilnacipran SR relative to placebo was 6 for response and 5 for remission, indicating that clearly clinically relevant outcomes occurred with levomilnacipran SR treatment.
A 10% difference in remission rates between SSRIs (35%) and placebo (25%) has been reported in a meta-analysis.21 In another meta-analysis,22 venlafaxine, the most extensively studied SNRI, showed a 13% difference in remission versus placebo (P < .001), with an NNT of 8. In the present study, the absolute difference for levomilnacipran SR versus placebo was 20% when measured by the MADRS and 13% when measured by the HDRS17, which demonstrates robust antidepressant effect leading to remission in a single, short-term study.
The large treatment effect on the primary outcome parameter was supported by significant outcomes in favor of levomilnacipran SR on all secondary outcomes and measures of clinical relevance such as NNT analyses and remission rates. The overtly positive nature of these results in a single, placebo-controlled study is an uncommon occurrence, which raises the question of whether this population was unusually responsive or levomilnacipran SR is potentially associated with superior efficacy. The lack of a reference antidepressant is a limitation of this study, which makes it impossible to address this question directly.
The inclusion (eg, minimum HDRS17 baseline score > 22) and exclusion (eg, the presence of comorbid anxiety disorders as required by regulatory authorities) criteria limit the ability to generalize these findings to routine clinical practice. The use of low-dose hypnotic and anxiolytic medications by patients in both treatment groups was allowed under certain circumstances, which may have further diminished the effects of anxious features and introduced uncertainties regarding the interpretation of some results. For example, concomitant low-dose hypnotic/anxiolytic use may have made it more difficult to demonstrate a drug-placebo difference; conversely, it may have reduced the dropout rate and allowed levomilnacipran SR time to show an effect compared with placebo. Use of hypnotics and anxiolytics, as well as the low level of baseline anxiety in the study population, may be additional study limitations.
Antidepressant efficacy potential is frequently lost as a result of poor tolerability and high levels of treatment discontinuation and noncompliance. In a meta-analysis23 comparing efficacy and acceptability of second-generation antidepressants, venlafaxine, escitalopram, and sertraline were among the more efficacious agents, but significantly more discontinuations for venlafaxine indicated a lower level of acceptability. In the present study, although the number of AEs reported was greater with levomilnacipran SR relative to placebo, the percentage of levomilnacipran SR dropouts (20.2%) was numerically lower than that of placebo dropouts (24.9%), suggesting good overall tolerability. Mean heart rate increase was more common in levomilnacipran SR patients than placebo, most likely due to increased noradrenergic effects.
The favorable tolerability profile of levomilnacipran SR may relate to the 2-fold greater potency for NE reuptake inhibition relative to 5-HT reuptake inhibition. By comparison, venlafaxine and duloxetine have markedly stronger preference for 5-HT reuptake inhibition, with approximately 30-fold and 10-fold greater potency for 5-HT versus NE reuptake inhibition, respectively.3 To achieve adequate norepinephrine reuptake inhibition, therapeutic doses of these agents need to be raised to the level at which potentially excessive serotonin reuptake inhibition may occur; this is associated with AEs such as nausea, sexual dysfunction, and discontinuation symptoms, which are commonly observed with venlafaxine and duloxetine.
Levomilnacipran is the more active enantiomer of milnacipran, an SNRI that is approved only for the treatment of fibromyalgia in the United States. On the basis of
double-blind trials versus placebo, tricyclic antidepressants, or SSRIs,24–29 twice-daily milnacipran has shown efficacy in major depressive episodes and is approved for the treatment of depression in several European countries. Milnacipran studies in depression were conducted a decade ago, and no head-to-head trials have been performed; as such, no valid comparison between levomilnacipran SR and milnacipran data can be made.
CONCLUSION
This first placebo-controlled study provides robust evidence that levomilnacipran SR had significant efficacy across all evaluated parameters and a good safety and tolerability profile. These results suggest that levomilnacipran SR will be a welcome addition to the antidepressant armamentarium. Further studies to support the present findings are ongoing.
Drug names: diazepam (Diastat, Valium, and others), duloxetine (Cymbalta), escitalopram (Lexapro and others), milnacipran (Savella), sertraline (Zoloft and others), venlafaxine (Effexor and others), zolpidem (Ambien, Edluar,
and others).
Author affiliations: Imperial College School of Medicine, University of London, London, United Kingdom (Dr Montgomery); Pierre Fabre Médicament, Toulouse, France (Dr Mansuy); Prescott Medical Communications Group, Chicago, Illinois (Dr Ruth); Forest Research Institute, Jersey City, New Jersey (Drs Bose, H. Li, and D. Li). Dr H. Li is
now employed by Novartis Pharmaceutical Corporation, East Hanover,
New Jersey.
Potential conflicts of interest: In the past year, Dr Montgomery has been a consultant to Forest, Lundbeck, Pierre Fabre, Richter, Servier, and Takeda
and a speaker or advisory board member for AstraZeneca, Lundbeck, and Pierre Fabre. Dr Mansuy is an employee of Pierre Fabre Médicament. Dr Ruth is an employee of Prescott Medical Communications Group, a contractor for Forest Research Institute. Drs Bose and D. Li are employees
of Forest Research Institute; at the time of the study, Dr H. Li was an employee of Forest Research Institute.
Funding/support: Supported by funding from Forest Research Institute, a subsidiary of Forest Laboratories Inc (New York, New York), and Pierre Fabre Médicament (Toulouse, France). Forest Laboratories, Inc was involved in the study design; collection (via contracted clinical investigator sites), analysis, and interpretation of data; and the decision to present these results.
Previous presentations: American College of Neuropsychopharmacology, December 6–10, 2009, Hollywood, Florida ▪ American Psychiatric Association, May 22–26, 2010, New Orleans, Louisiana ▪ Collegium Internationale Neuro-Psychopharmacologicum, June 6–10, 2010, Hong Kong, China ▪ New Clinical Drug Evaluation Unit, June 14–17, 2010, Boca Raton, Florida ▪ European College of Neuropsychopharmacology, August 28–September 1, 2010, Amsterdam, The Netherlands ▪ Institute of Psychiatric Services Annual Meeting, October 14–17, 2010, Boston, Massachusetts ▪ American Psychiatric Association, May 14–18, 2011, Honolulu, Hawaii ▪ and New Clinical Drug Evaluation Unit, June 13–16, 2011, Boca Raton, Florida.
Acknowledgments: The authors acknowledge the late Yves Lecrubier, MD, of the French National Institutes of Health and Medical Research (INSERM), Hôpital Pitié-Salpêtrière, Paris, France, the principal investigator for this study until his untimely illness made it impossible for him to continue. Writing assistance and editorial support for the preparation of this manuscript were provided by Carol Dyer, MS, of Prescott Medical Communications Group, Chicago, Illinois, a contractor of Forest Research Institute.
REFERENCES
1. Murray CJ, Lopez AD. Global mortality, disability, and the contribution
of risk factors: Global Burden of Disease Study. Lancet. 1997;349(9063):1436–1442. doi:10.1016/S0140-6736(96)07495-8 PubMed
2. Blier P, Saint-André E, Hébert C, et al. Effects of different doses of
venlafaxine on serotonin and norepinephrine reuptake in healthy volunteers. Int J Neuropsychopharmacol. 2007;10(1):41–50. doi:10.1017/S1461145705006395 PubMed
3. Stahl SM, Grady MM, Moret C, et al. SNRIs: their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005;10(9):732–747. PubMed
4. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382–389. doi:10.1192/bjp.134.4.382 PubMed
5. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
6. Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(suppl 20):22–33, quiz 34–57. PubMed
7. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56–62. doi:10.1136/jnnp.23.1.56 PubMed
8. Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability.
Int Clin Psychopharmacol. 1996;11(suppl 3):89–95. doi:10.1097/00004850-199606003-00015 PubMed
9. Guy W. The clinician global severity and impression scales. ECDEU Assessment Manual for Psychopharmacology. Department of Health, Education, and Welfare publication no. 76-338. Rockville, MD: National Institute of Mental Health; 1976; 218–222.
10. Lipman R, Covi L. Outpatient treatment of neurotic depression: medication and group psychotherapy. In: Spitzer R, Klein D, eds. Evaluation of Psychological Therapies. Baltimore, MD: John Hopkins University Press;
1976: 178–218.
11. Altman DG. Confidence intervals for the number needed to treat. BMJ. 1998;317(7168):1309–1312. doi:10.1136/bmj.317.7168.1309 PubMed
12. Harmonised Tripartite Guideline ICH. The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. May 12, 2005. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf. Accessed
October 22, 2012.
13. Khan A, Leventhal RM, Khan SR, et al. Severity of depression and
response to antidepressants and placebo: an analysis of the Food and Drug Administration database. J Clin Psychopharmacol. 2002;22(1):40–45. doi:10.1097/00004714-200202000-00007 PubMed
14. Melander H, Salmonson T, Abadie E, et al. A regulatory Apologia—a review of placebo-controlled studies in regulatory submissions of new-generation antidepressants. Eur Neuropsychopharmacol. 2008;18(9):623–627. doi:10.1016/j.euroneuro.2008.06.003 PubMed
15. Kirsch I. The emperor’s new drugs: an analysis of antidepressant medication data submitted to the US Food and Drug Administration. Prev Treat. 2002;5(article 23):1–11. http://alphachoices.com/repository/assets/pdf/EmperorsNewDrugs.pdf. Accessed January 30, 2012.
16. Hirschfeld RM, Montgomery SA, Keller MB, et al. Social functioning in depression: a review. J Clin Psychiatry. 2000;61(4):268–275. doi:10.4088/JCP.v61n0405 PubMed
17. Israel JA. Remission in depression: definition and initial treatment approaches. J Psychopharmacol. 2006;20(suppl):5–10. doi:10.1177/1359786806064306 PubMed
18. Thase ME. The small specific effects of antidepressants in clinical trials: what do they mean to psychiatrists? Curr Psychiatry Rep. 2011;13(6):476–482. doi:10.1007/s11920-011-0235-x PubMed
19. Montgomery SA, Möller HJ. Is the significant superiority of escitalopram compared with other antidepressants clinically relevant? Int Clin Psychopharmacol. 2009;24(3):111–118. doi:10.1097/YIC.0b013e32832a8eb2 PubMed
20. Kraemer HC, Kupfer DJ. Size of treatment effects and their importance to clinical research and practice. Biol Psychiatry. 2006;59(11):990–996. doi:10.1016/j.biopsych.2005.09.014 PubMed
21. Thase ME. Are SNRIs more effective than SSRIs? a review of the current
state of the controversy. Psychopharmacol Bull. 2008;41(2):58–85. PubMed
22. Nemeroff CB, Entsuah R, Benattia I, et al. Comprehensive analysis of remission (COMPARE) with venlafaxine versus SSRIs. Biol Psychiatry. 2008;63(4):424–434. doi:10.1016/j.biopsych.2007.06.027 PubMed
23. Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373(9665):746–758. doi:10.1016/S0140-6736(09)60046-5 PubMed
24. Guelfi JD, Ansseau M, Corruble E, et al. A double-blind comparison of the efficacy and safety of milnacipran and fluoxetine in depressed inpatients.
Int Clin Psychopharmacol. 1998;13(3):121–128. doi:10.1097/00004850-199805000-00005 PubMed
25. Kasper S, Pletan Y, Solles A, et al. Comparative studies with milnacipran and tricyclic antidepressants in the treatment of patients with major depression:
a summary of clinical trial results. Int Clin Psychopharmacol. 1996;
11(suppl 4):35–39. doi:10.1097/00004850-199609004-00005 PubMed
26. Lecrubier Y, Pletan Y, Solles A, et al. Clinical efficacy of milnacipran:
placebo-controlled trials. Int Clin Psychopharmacol. 1996;11(suppl 4):29–33. doi:10.1097/00004850-199609004-00004 PubMed
27. Lopez-Ibor J, Guelfi JD, Pletan Y, et al. Milnacipran and selective serotonin reuptake inhibitors in major depression. Int Clin Psychopharmacol. 1996;11(suppl 4):41–46. doi:10.1097/00004850-199609004-00006 PubMed
28. Montgomery SA, Prost JF, Solles A, et al. Efficacy and tolerability of milnacipran: an overview. Int Clin Psychopharmacol. 1996;11(suppl 4):47–51. doi:10.1097/00004850-199609004-00007 PubMed
29. Van Amerongen AP, Ferrey G, Tournoux A. A randomised, double-blind comparison of milnacipran and imipramine in the treatment of depression. J Affect Disord. 2002;72(1):21–31. doi:10.1016/S0165-0327(01)00422-0 PubMed
This PDF is free for all visitors!
![]() Save
Save
![]() Share
Share
![]() Cite
Cite