Context: Vortioxetine (Lu AA21004) is an antidepressant with a mechanism of action thought to be related to a combination of 2 pharmacologic actions: direct modulation of several receptors and inhibition of the serotonin transporter.
Objective: To evaluate the efficacy of vortioxetine 10 and 20 mg once daily in outpatients with major depressive disorder.
Design, Setting, and Participants: This 8-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study was conducted from July 2010 to January 2012 among adults with a primary diagnosis of recurrent major depressive disorder (DSM-IV-TR).
Intervention: Eligible subjects were randomized in 1:1:1 ratio to 1 of 3 treatment arms: vortioxetine 10 mg, vortioxetine 20 mg, or placebo once daily for 8 weeks. Subjects who completed the 8-week trial entered a 2-week blinded discontinuation period to assess potential discontinuation symptoms.
Main Outcome Measure: The primary endpoint was the least squares mean change in Montgomery-Asberg Depression Rating Scale (MADRS) total score from baseline. Key secondary outcomes were analyzed in the following prespecified sequential order: MADRS response (≥ 50% decrease from baseline in total score), Clinical Global Impressions-Improvement score, change from baseline in MADRS total score in subjects with baseline Hamilton Anxiety Rating Scale score ≥ 20, MADRS remission (total score ≤ 10), and change from baseline in Sheehan Disability Scale total score (all at week 8).
Results: A total of 462 subjects were randomized to placebo (n = 157), vortioxetine 10 mg (n = 155), and vortioxetine 20 mg (n = 150). Mean (SE) reductions from baseline in MADRS total score (week 8) were -10.77 (± 0.807), -12.96 (± 0.832), and -14.41 (± 0.845) for the placebo, vortioxetine 10 mg (P = .058 vs placebo), and vortioxetine 20 mg (P = .002 vs placebo) groups. MADRS response/remission was achieved in 28.4%/14.2%, 33.8%/21.4%, and 39.2%/22.3% of subjects, respectively, in the 3 groups. Only MADRS response for vortioxetine 20 mg significantly separated from placebo (P = .044). Treatment was well tolerated, with the most frequently reported adverse events consisting of nausea, headache, diarrhea, and dizziness.
Conclusions: Vortioxetine 20 mg significantly reduced MADRS total score at 8 weeks in this study population. Overall, vortioxetine was well tolerated in this study.
Trial Registration: ClinicalTrials.gov identifier: NCT01163266
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
This work may not be copied, distributed, displayed, published, reproduced, transmitted, modified, posted, sold, licensed, or used for commercial purposes.
By downloading this file, you are agreeing to the publisher’s Terms & Conditions.
A Randomized, Double-Blind, Placebo-Controlled Study
of the Efficacy and Safety of Vortioxetine 10 mg and 20 mg
in Adults With Major Depressive Disorder
ABSTRACT
Context: Vortioxetine (Lu AA21004) is an antidepressant with a mechanism of action thought to be related to a combination of 2 pharmacologic actions: direct modulation of several receptors and inhibition of the serotonin transporter.
Objective: To evaluate the efficacy of vortioxetine 10 and 20 mg once daily in outpatients with major depressive disorder.
Design, Setting, and Participants: This 8-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study was conducted from July 2010 to January 2012 among adults with a primary diagnosis of recurrent major depressive disorder (DSM-IV-TR).
Intervention: Eligible subjects were randomized in 1:1:1 ratio to 1 of 3 treatment arms: vortioxetine 10 mg, vortioxetine 20 mg, or placebo once daily for 8 weeks. Subjects who completed the 8-week trial entered a 2-week blinded discontinuation period to assess potential discontinuation symptoms.
Main Outcome Measure: The primary endpoint was the least squares mean change in Montgomery-Asberg Depression Rating Scale (MADRS) total score from baseline. Key secondary outcomes were analyzed in the following prespecified sequential order: MADRS response (≥ 50% decrease from baseline in total score), Clinical Global Impressions-Improvement score, change from baseline in MADRS total score in subjects with baseline Hamilton Anxiety Rating Scale score ≥ 20, MADRS remission (total score ≤ 10), and change from baseline in Sheehan Disability Scale total score (all at week 8).
Results: A total of 462 subjects were randomized to placebo (n = 157), vortioxetine 10 mg (n = 155), and vortioxetine 20 mg (n = 150). Mean (SE) reductions from baseline in MADRS total score (week 8) were –10.77 (± 0.807), –12.96 (± 0.832), and –14.41 (± 0.845) for the placebo, vortioxetine 10 mg (P = .058 vs placebo), and vortioxetine 20 mg (P = .002 vs placebo) groups. MADRS response/remission was achieved in 28.4%/14.2%, 33.8%/21.4%, and 39.2%/22.3% of subjects, respectively, in the 3 groups. Only MADRS response for vortioxetine 20 mg significantly separated from placebo (P = .044). Treatment was well tolerated, with the most frequently reported adverse events consisting of nausea, headache, diarrhea, and dizziness.
Conclusions: Vortioxetine 20 mg significantly reduced MADRS total score at 8 weeks in this study population. Overall, vortioxetine was well tolerated in this study.
Trial Registration: ClinicalTrials.gov identifier: NCT01163266
J Clin Psychiatry 2015;76(5):575–582
© Copyright 2015 Physicians Postgraduate Press, Inc.
Submitted: June 23, 2014; accepted January 15, 2015
(doi:10.4088/JCP.14m09335).
Corresponding author: Madhukar H. Trivedi, MD, 6363 Forest Park Rd, Ste 13.354, Dallas, TX 75390-9119 ([email protected]).
Major depressive disorder (MDD), a common, chronic disorder, is the fourth leading cause of overall disease burden.1 In developed countries, depression is the leading cause of lost disability-adjusted life-years.1 Depression is also associated with an increased risk of suicide.2,3
Despite availability of several antidepressant classes, data suggest that ~50% of patients do not respond to treatment and approximately two thirds fail to achieve remission.4–6 In addition, relapse is common.7 Furthermore, antidepressant treatment is associated with adverse events (AEs), including weight changes, sexual dysfunction, and insomnia, that can affect patient compliance with therapy.8–10
Vortioxetine (Lu AA21004) is approved in the United States and Europe for the treatment of MDD with a mechanism of action thought to be related to its multimodal activity. It combines 2 pharmacologic actions: direct modulation of serotonin receptor activity and inhibition of the serotonin transporter. In vitro studies indicate that vortioxetine is a 5-HT3, 5-HT7, and 5-HT1D receptor antagonist; a 5-HT1A receptor agonist;
a 5-HT1B receptor partial agonist; and an inhibitor of
the 5-HT transporter.11,12 The precise contribution of the individual targets to the observed pharmacodynamic profile remains unclear. However, data from serotonergic receptor and transporter occupancy studies, coupled with neuronal firing and microdialysis studies in rats, suggest that these targets interact in a complex fashion, leading to modulation of neurotransmission in several systems, including serotonin, norepinephrine, dopamine, histamine, glutamate, and acetylcholine systems within the rat forebrain.11–13
Previous clinical trials have demonstrated efficacy for vortioxetine 5–20 mg in reducing depression symptoms, with the majority of studies evaluating the lower doses (5–10 mg).14–18 The primary objective of this study was to evaluate the efficacy of vortioxetine 10 and 20 mg once daily compared with placebo in subjects with MDD, as assessed by the Montgomery-Asberg Depression Rating Scale (MADRS).19
METHOD
Study Design
This was a phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group study conducted from July 2010 through January 2012 at 37 sites in the United States. Eligible subjects were randomized in a 1:1:1 ratio to 1 of 3 treatment arms: vortioxetine 10 mg, vortioxetine 20 mg, or placebo once daily for 8 weeks. Subjects assigned to the vortioxetine 20 mg group received a 10 mg dose for the first week and then 20 mg for the remaining 7 weeks. Those assigned to the vortioxetine 10 mg or placebo groups received vortioxetine 10 mg or placebo for the entire 8 weeks. All medication was dispensed weekly in identical blister packs to maintain blinding. Subjects who completed the 8-week trial entered a 2-week blinded discontinuation period to assess potential discontinuation symptoms. The study was conducted in accordance with the Declaration of Helsinki, International Conference on Harmonization Guidelines
for Good Clinical Practice, and was approved by the institutional review boards of participating centers. All subjects provided written informed consent prior to study entry. The study is registered at ClinicalTrials.gov (identifier: NCT01163266).
Subjects
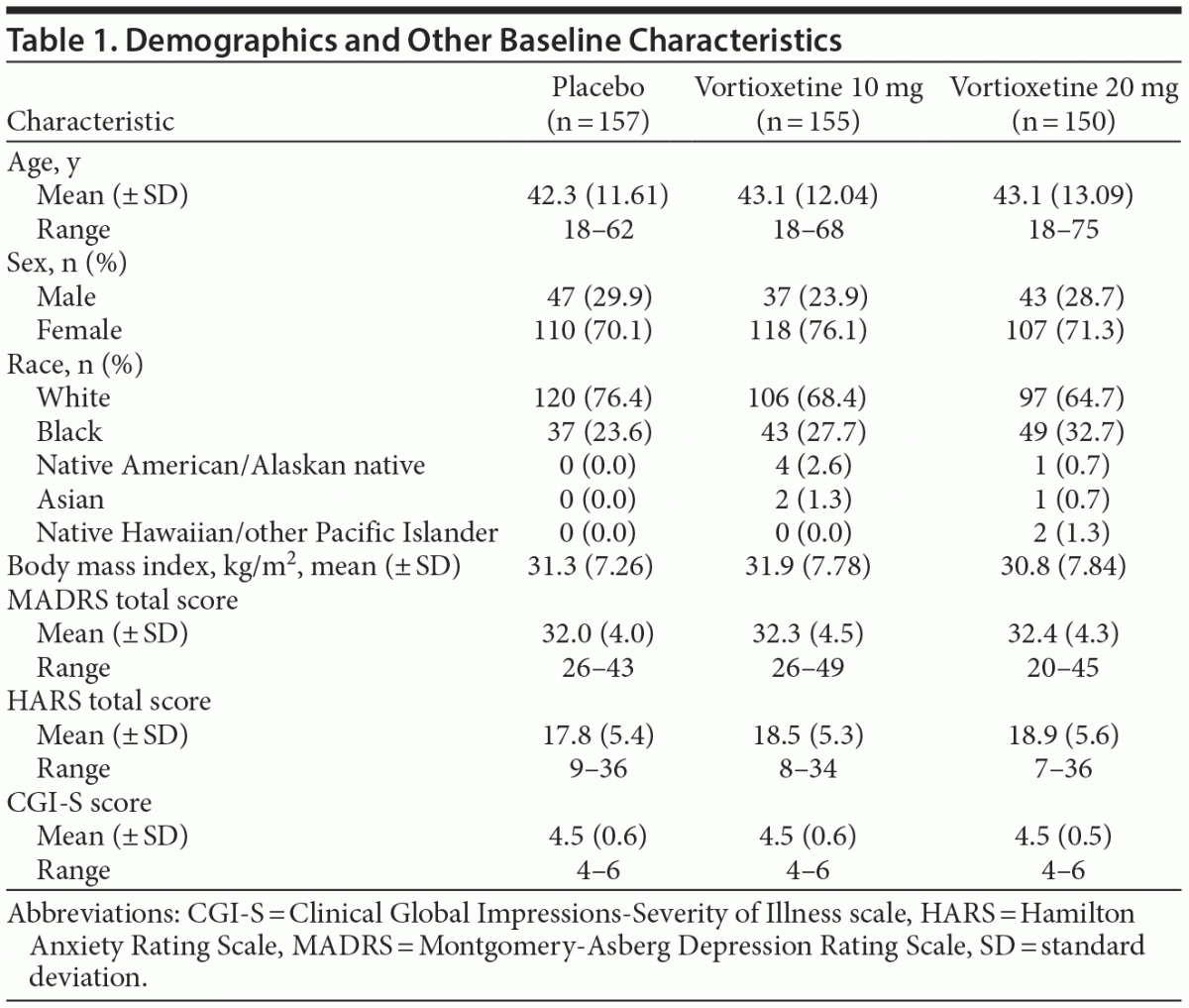
Men and women aged 18–75 years (inclusive) with a primary diagnosis of recurrent MDD (classification code 296.3x), as defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision,20 were eligible for enrollment. Other inclusion criteria included the presence of a current major depressive episode confirmed by the Structured Clinical Interview for DSM Disorders with a duration of ≥ 3 months, MADRS total score ≥ 26, and Clinical Global Impressions-Severity of Illness (CGI-S)21 score ≥ 4 at screening and baseline visits.
Exclusion criteria included current psychiatric disorders or past history of psychotic disorder, a current diagnosis of alcohol or other substance abuse or dependence, and the presence or history of a significant neurologic disorder, neurodegenerative disorder, or clinically unstable medical illness. Subjects with current depressive symptoms considered to be resistant to 2 adequate antidepressant treatments for ≥ 6 weeks or a significant suicide risk were excluded. All subjects were required to have a 2-week (or longer depending on drug half-life) washout period for any psychoactive medications prior to screening.
Assessments
Subjects were assessed at screening and baseline, weekly during the first 2 weeks of treatment, and then every 2 weeks up to the 8-week study end. Efficacy assessments included the MADRS, Clinical Global Impressions-Improvement scale (CGI-I)21 and CGI-S, Hamilton Anxiety Rating Scale (HARS),22 and Sheehan Disability Scale (SDS).23 With the exception of the SDS, which was assessed at baseline and weeks 6 and 8, efficacy assessments were performed at each study visit. The primary endpoint was change from baseline in the MADRS total score after 8 weeks. Key secondary endpoints, assessed in a hierarchical manner, included MADRS response (ie, ≥ 50% decrease in MADRS total score from baseline at week 8), mean CGI-I score at week 8, change from baseline in MADRS total score at week 8 in subjects with baseline HARS total score ≥ 20, MADRS remission (MADRS total score ≤ 10 at week 8), and change from baseline in SDS total score at week 8. Prespecified subgroup analyses by age, sex, race, and baseline severity of depression and anxiety symptoms were also conducted for the primary endpoint.
Safety was assessed via AEs (at each study visit), clinical laboratory results (baseline and week 4), vital signs (at each study visit), electrocardiogram (screening and week 4), weight (screening/baseline and week 4), and physical examination findings (screening and completion). AEs were evaluated for severity (mild, moderate, or severe) and causal relationship to study drug (ie, probable, possible, or not related). Suicidal ideation and behavior (SIB) was assessed using the Columbia-Suicide Severity Rating Scale (C-SSRS),24 and sexual function was evaluated using the Arizona Sexual Experience Scale (ASEX)25 at each study visit. Discontinuation symptoms were assessed weekly beginning at week 8 (baseline) and for 2 weeks following discontinuation using the Discontinuation-Emergent Signs and Symptoms (DESS)26 scale, as well as spontaneously reported AEs.
Statistical Analysis
Subjects who were randomized and received ≥ 1 dose of study drug and had ≥ 1 postbaseline value for primary efficacy assessment were included in the analysis set. Assuming a standard deviation of 9.5 for the change from baseline in MADRS total score, a total of ~450 subjects was considered sufficient to achieve ≥ 80% power to detect a difference of 3.5 points on the MADRS between a vortioxetine dose and placebo by a 2-sample t test with a 2-sided significance level of .025.
The primary endpoint (least squares [LS] mean change in MADRS total score from baseline) was based on a mixed model for repeated measures analysis of covariance (ANCOVA) with treatment, center, week, treatment-by-week interaction, and baseline MADRS total score by week as fixed effects. As supportive analysis, the primary endpoint was also analyzed using ANCOVA with treatment and center as fixed factors and baseline MADRS total score as covariate and using last observation carried forward (LOCF) and observed case (OC) methods.
Secondary variables (HARS, CGI-S, CGI-I) were analyzed in a similar manner to the primary endpoint. Response and remission rates were analyzed at all time points by logistic regression adjusting for baseline score and treatment by both LOCF and OC methods. All statistical tests were 2-sided at a 5% significance level. To control for a 2-sided type I error, secondary efficacy endpoints were tested for each dose in the following sequential order at a .025 significance level: change from baseline in MADRS total score, MADRS responders, mean CGI-I score, change from baseline in MADRS total score in subjects with baseline HARS score ≥ 20, MADRS remission, and change from baseline in SDS total score (all at week 8). As soon as an endpoint was nonsignificant at .025, the formal testing procedure stopped for all subsequent endpoints for that dose, and all P values < .05 for that dose were considered nominal and described as separated from placebo.
The primary ASEX analysis was the assessment of subjects who did not have sexual dysfunction at baseline and developed it at any time during the study period. Each subject not having sexual dysfunction at baseline was evaluated for a shift to having sexual dysfunction at any visit during treatment using the following definition: (1) an ASEX total score ≥ 19, (2) a score of ≥ 5 on any item, or (3) a score of ≥ 4 on any 3 items. For any other scoring, a subject was considered not to have sexual dysfunction. Two-sided tests with 95% confidence intervals constructed using the normal approximation to binomial were used to analyze the differences between the incidence rates for vortioxetine-treated (pooled) and placebo-treated subjects.
SIB was prospectively monitored using C-SSRS. The report was considered positive if the subject reported any of the following: active suicidal ideation with some intent to act without specific plan, active suicidal ideation with specific plan and intent, interrupted/aborted suicide attempt, preparatory acts/behavior, actual attempt, or completed suicide.
At completion of the 8-week treatment period, subjects in the vortioxetine groups were abruptly switched to placebo, while subjects in the placebo group remained on placebo during the discontinuation period. In subjects who completed the 8-week double-blind period, potential discontinuation symptoms were assessed using the DESS scale, which was administered during a blinded 2-week discontinuation period (weeks 9 and 10). Descriptive statistics were reported for AEs, vital signs, weight, laboratory values, electrocardiogram, and physical examination findings.
RESULTS
Subjects
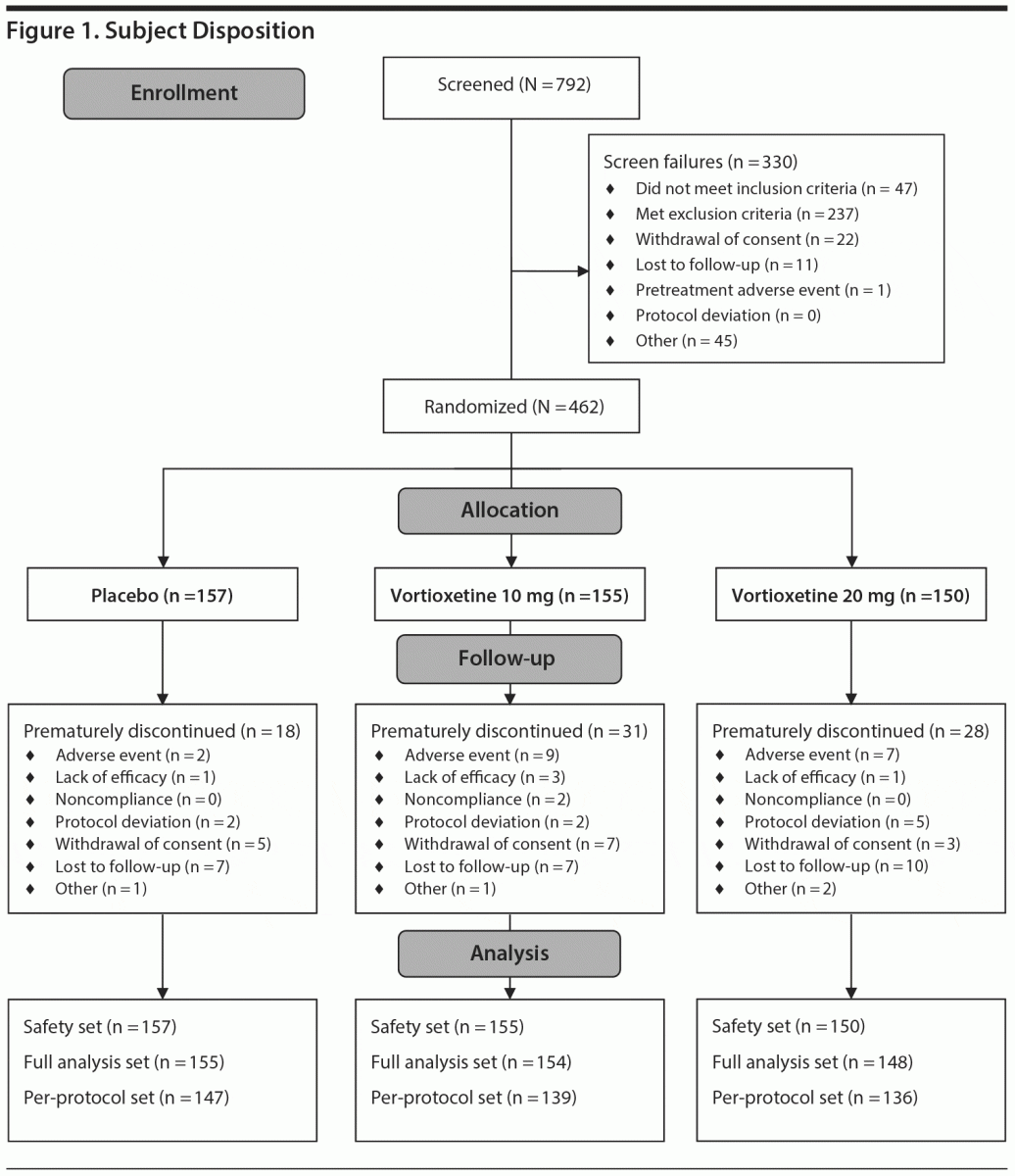
Of 792 subjects screened, 462 were randomized and received the study drug (placebo, n = 157; vortioxetine 10 mg, n = 155; vortioxetine 20 mg, n = 150). Seventy-seven subjects prematurely discontinued treatment, with the most common reasons being lost to follow-up (n = 24), AE (n = 18), withdrawal of consent (n = 15), or protocol deviation (n = 9). All 462 subjects were included in the safety set, and 457 (98.9%) were included in the full analysis set, with 155/157 (98.7%) in the placebo group, 154/155 (99.4%) in the vortioxetine 10 mg group, and 148/150 (98.7%) in the vortioxetine 20 mg group (Figure 1). Subject characteristics are summarized in Table 1. Mean MADRS total scores at baseline ranged from 32.0 to 32.4 across treatment groups, and the mean CGI-S score was 4.5 in all treatment groups, indicating that, overall, subjects had moderate-to-severe MDD and were moderately to markedly ill.
Efficacy
In the primary efficacy analysis (LS mean change from baseline in MADRS total score at week 8), vortioxetine 20 mg was statistically significantly superior to placebo (Table 2). The mean difference between vortioxetine 20 mg and placebo for MADRS total score was –3.64 (SE ± 1.161; P = .002). The difference between vortioxetine 10 mg and placebo in MADRS change from baseline did not reach significance at week 8 (P = .058). The mean changes from baseline in MADRS total score over the study course are illustrated in Figure 2. Vortioxetine 20 mg separated from placebo at week 4 and remained separated at weeks 6 and 8. The vortioxetine 10 mg dose also separated from placebo at weeks 4 and 6 but not at week 8.
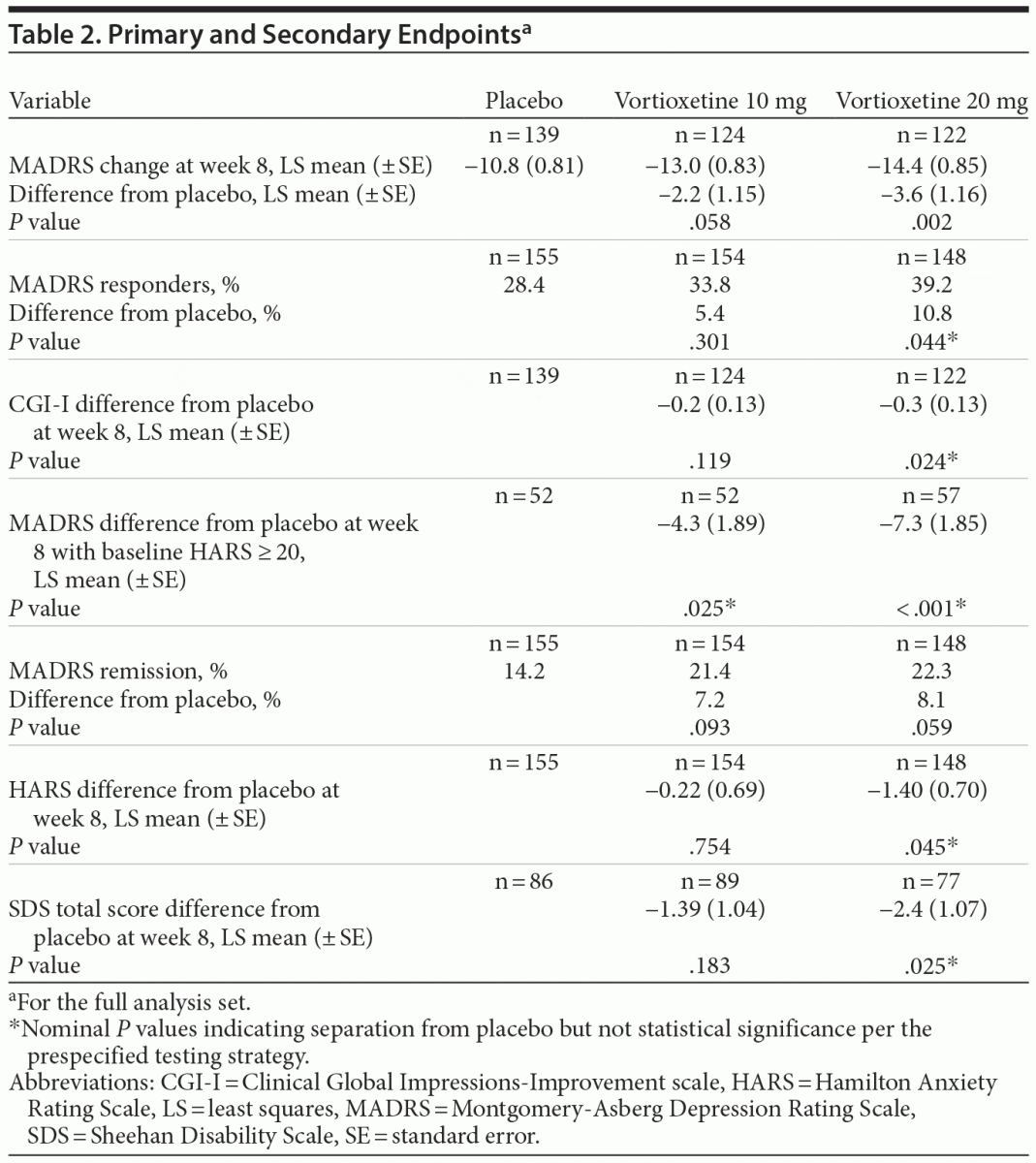
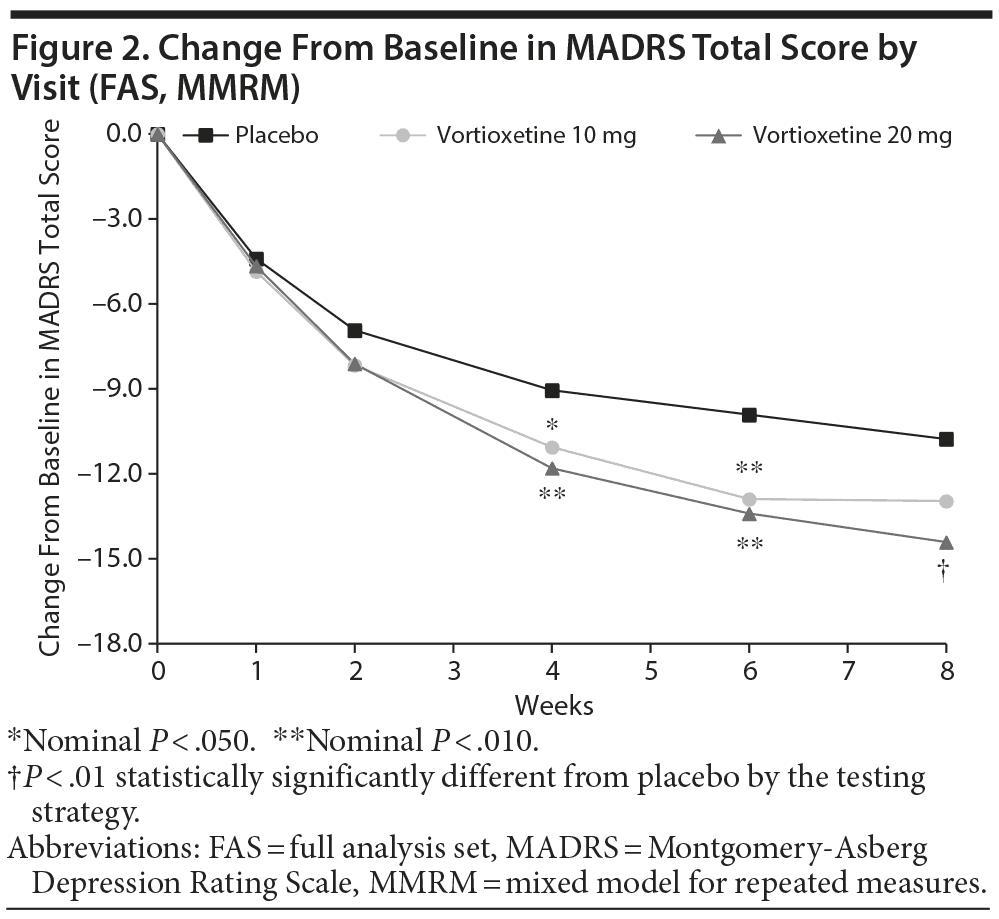
There was no apparent difference in efficacy (LS mean MADRS total score change from baseline at week 8) when subjects were stratified by age (≤ 55 years: vortioxetine 10 mg −12.61, vortioxetine 20 mg −14.57; > 55 years: 10 mg −13.62, 20 mg −13.00), sex (male: 10 mg −10.78, 20 mg −14.94; female: 10 mg −13.75, 20 mg −14.13), or race (white: 10 mg −13.73, 20 mg −14.74; black: 10 mg −12.26, 20 mg −14.62). However, women tended to have a higher placebo response versus men, and men tended to have more of a dose-response effect (ie, greater response at 20 vs 10 mg) than did women. In addition, subjects with higher baseline MADRS (> 32) and HARS (≥ 20) scores tended to have greater efficacy than those with lower baseline scores. Notably, subjects with higher baseline HARS scores had greater reductions from baseline in MADRS total score in both vortioxetine dose groups (Table 2). In this group, a greater magnitude of the effect was observed with vortioxetine 20 mg versus vortioxetine 10 mg. For subjects with moderately severe depression (ie, MADRS > 32), vortioxetine 10 mg (−16.80) was similar in efficacy to the 20 mg dose (−15.64).
MADRS response at 8 weeks (≥ 50% decrease from baseline in MADRS total score) was achieved in 33.8%, 39.2%, and 28.4% of subjects in the vortioxetine 10 mg, 20 mg, and placebo groups, respectively (P = .301 [10 mg vs placebo]; P = .044 [20 mg vs placebo]). Since the difference did not reach the predefined level of statistical significance (.025), the hierarchical testing strategy was stopped, and all subsequent P values (< .05) were considered nominal and not statistically significant. At week 8, CGI-I and SDS differences from placebo were nominally significant for vortioxetine 20 mg, while MADRS differences from placebo among those with a baseline HARS score ≥ 20 were nominally significant for both vortioxetine doses. MADRS remission (MADRS total score ≤ 10 at week 8) was achieved in 21.4%, 22.3%, and 14.2% of subjects in the vortioxetine 10 mg, vortioxetine 20 mg, and placebo groups, respectively, but neither vortioxetine dose separated from placebo.
The mean change from baseline in HARS total score is shown in Table 2. Vortioxetine 20 mg attained nominal difference versus placebo at week 8, with a mean reduction of 7.6 points versus a reduction of 6.2 points in the placebo group at week 8. Vortioxetine 20 mg was also associated with a greater reduction in CGI-I score versus placebo at weeks 2, 4, 6, and 8, whereas it was superior to placebo at weeks 4 and 6 in CGI-S score. In addition, vortioxetine 20 mg was associated with a greater decline in SDS total scores at week 8 versus placebo (–8.3 vs –5.9; nominal P = .025).
Safety
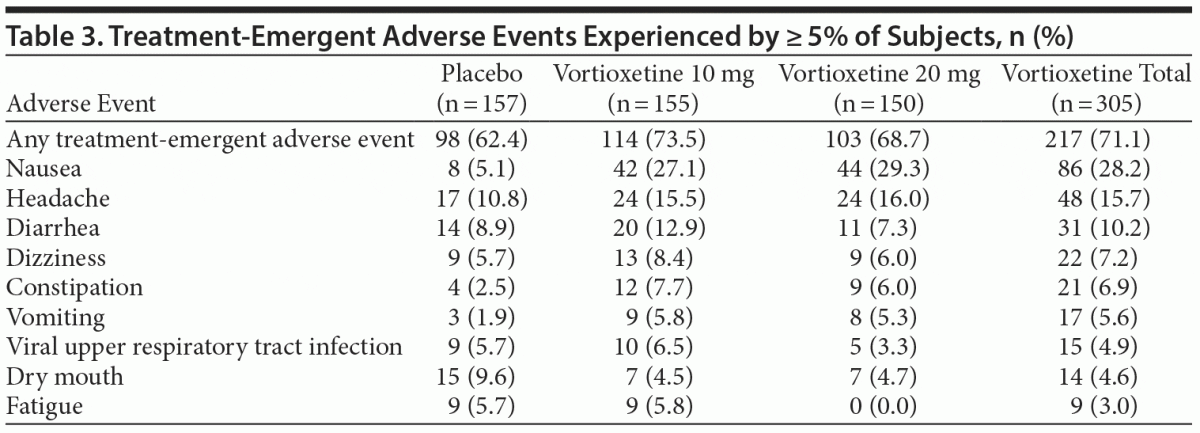
Treatment-emergent AE (TEAE) rates were 73.5%, 68.7%, and 62.4% in the vortioxetine 10 mg, vortioxetine 20 mg, and placebo groups, respectively (Table 3). The most frequently reported AEs (≥ 5%) in the vortioxetine treatment groups were nausea, headache, diarrhea, dizziness, constipation, vomiting, viral upper respiratory tract infections, and fatigue. Of these, nausea was the most common AE in the vortioxetine treatment groups (28.2% overall) versus 5.1% for placebo-treated patients. There were no differences in incidence of insomnia between placebo (n = 6) and either vortioxetine group (n = 6, vortioxetine 10 mg; n = 3, vortioxetine 20 mg). Most AEs were mild or moderate in severity. There were 2 serious AEs, both in the vortioxetine 10 mg treatment group: 1 case of kidney infection and 1 suicide attempt.
Seventeen subjects had TEAEs leading to study discontinuation: 8 (5.2%), 7 (4.7%), and 2 (1.3%) subjects in the vortioxetine 10 mg, vortioxetine 20 mg, and placebo groups, respectively. In subjects who received vortioxetine 10 or 20 mg, the most common AEs leading to treatment discontinuation were nausea (n = 5), headache (n = 2), and fatigue (n = 2).
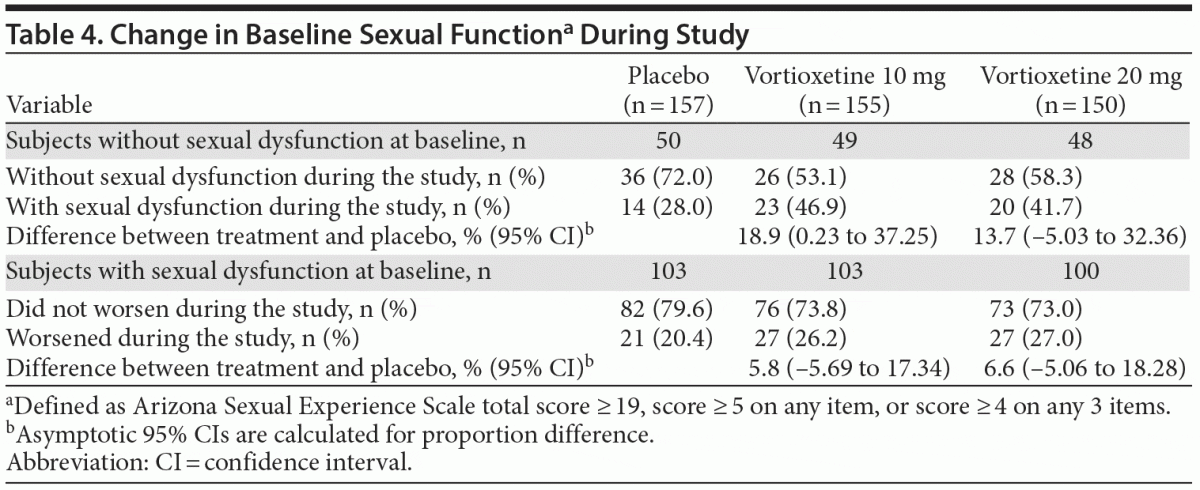
Approximately one third of subjects in each treatment group had no sexual dysfunction at baseline as assessed by ASEX (Table 4). In the vortioxetine 10 mg group, subjects without sexual dysfunction at baseline had an 18.9% higher incidence of sexual dysfunction during the study versus placebo. In the vortioxetine 20 mg group, subjects without sexual dysfunction at baseline had a 13.7% higher rate of sexual dysfunction during the study than placebo. There were no differences from placebo in the change from baseline of ASEX total score at week 8 for the vortioxetine 10 mg and 20 mg groups, with mean changes from baseline at week 8 of –0.99, –0.85, and –0.36 in the vortioxetine 10 mg, vortioxetine 20 mg, and placebo groups, respectively.
At the baseline (lifetime) assessment, the number of subjects with positive C-SSRS reports was similar across the vortioxetine 10 mg (16.8%), vortioxetine 20 mg (12.7%), and placebo (17.8%) groups. Shift analysis revealed no positive C-SSRS reports in the placebo or vortioxetine 20 mg groups. A severely depressed 45-year-old female subject in the vortioxetine 10 mg group reported suicidal ideation with intent to act and also made a suicide attempt. This serious event resulted in early discontinuation from the study, and the subject recovered.
No statistically significant differences in DESS total scores were observed between vortioxetine (10 and 20 mg) and placebo during the 2-week discontinuation period (weeks 9 and 10) following an abrupt discontinuation of vortioxetine at the end of week 8.
Changes in serum chemistry, hematology, vital signs, and electrocardiogram parameters were distributed evenly across treatment groups. Mean weight changes from baseline at week 8 were small in the 20 mg (–0.10 kg) and 10 mg (0.19 kg) groups, similar to that with placebo (0.46 kg).
DISCUSSION
The primary endpoint was met, supporting the efficacy of vortioxetine 20 mg in the treatment of MDD. For subjects receiving the 20 mg dose, the difference from placebo in the baseline-adjusted MADRS total score change was 3.6 points. In addition, response and remission were achieved in ~10% more subjects in the 20 mg dose group at week 8 (39.2% and 22.3%, respectively) versus placebo (28.4% and 14.2%, respectively), but this separation was not statistically significant. MADRS total scores for subjects in both vortioxetine groups began to demonstrate difference from placebo at week 2, and the values separated from placebo by week 4. Statistically significant separation for the 20 mg dose group was maintained through weeks 6 and 8. For the 10 mg dose group, statistically significant separation continued through week 6 (nominal P < .05). However, the separation from placebo was not maintained at week 8, as the mean MADRS declined with placebo while remaining steady in the vortioxetine 10 mg group. The efficacy of vortioxetine for reducing MADRS scores was evident across various demographic groups (age, sex, and race). In addition, vortioxetine was more efficacious among those with higher disease severity at baseline (ie, baseline MADRS > 32; baseline HARS ≥ 20). Overall efficacy of vortioxetine 20 mg was supported by improvement in anxiety symptoms (HARS), disability (SDS), and severity of illness (CGI-I) at the 8-week study endpoint.
Previous vortioxetine studies demonstrated positive improvements with 10 mg doses on multiple endpoints, so the reason for the lack of significant separation from placebo in the current study for the 10 mg dose is unknown.14–16 It is possible that overall difficulties in signal detection in mood disorder trials may be a factor. However, the significant separation from placebo with the vortioxetine 20 mg dose is consistent with previous studies and suggests that higher doses may potentially demonstrate greater improvements.17,18 This is supported by the known dose-response relationship that has been demonstrated for vortioxetine and MADRS total score.27
Vortioxetine was well tolerated, with a low incidence of AEs. Most events were mild to moderate in intensity and did not result in treatment discontinuation. The most commonly reported AEs (nausea, headache, diarrhea, dizziness, constipation, vomiting, viral upper respiratory tract infection, and fatigue) were consistent with those reported in other vortioxetine studies.14–18,28–30
Based on known antidepressant effects, the current study was designed to prospectively and systematically assess discontinuation symptoms and sexual functioning, as well as SIB, using specific scales. Antidepressants such as selective serotonin reuptake inhibitors are associated with a discontinuation syndrome characterized by nausea, lethargy, insomnia, vertigo, and sensory abnormalities.31,32 However, the potential discontinuation symptoms (as assessed by DESS) during the 2 weeks following abrupt cessation of vortioxetine treatment revealed no statistically significant difference in the vortioxetine treatment groups versus placebo. Changes in sexual functioning are often associated with psychological disorders including MDD. Effective treatment may mitigate these changes. However, treatment with a pharmacologic agent may also impact sexual desire and performance.33 Although the sexual dysfunction rate reported in the 10 mg group was uncharacteristically high versus other studies evaluating vortioxetine 10 mg,29,34 the rate in the 20 mg dose group was not significantly greater than in the placebo group. Furthermore, most subjects did not experience worsening of existing sexual dysfunction as assessed by the ASEX. The SIB incidence (as assessed by the C-SSRS) was low, with similar rates in the vortioxetine- and placebo-treated groups.
Limitations of this study include those common with most randomized clinical trials. The data from a clinical trial may be difficult to generalize to all MDD patients, since study subjects are not fully representative of those in real-world settings. Clinical trials use stringent inclusion/exclusion criteria to define a more pure MDD population than is seen in the real world, where patients often have medical and psychiatric comorbidities. The design of this trial also did not include the lower approved doses of vortioxetine, nor allow for flexible dosing, which would be more representative of real-world treatment, where the dose could be tailored to the patient for optimal efficacy and tolerability. In addition, multicenter trials with large numbers of sites may lead to higher variability in results, making separation from placebo more difficult.
CONCLUSION
In conclusion, vortioxetine 20 mg significantly reduced MADRS total score at 8 weeks in adults with MDD. Overall, vortioxetine was well tolerated in this study.
Drug names: vortioxetine (Brintellix).
Author affiliations: Takeda Development Center Americas, Deerfield, Illinois (Drs Mahableshwarkar, Serenko, and Chan and Ms Jacobsen); and UT Southwestern Medical Center, Dallas, Texas (Dr Trivedi).
Potential conflicts of interest: Ms Jacobsen and Dr Mahableshwarkar are employees of Takeda Development Center Americas. Dr Mahableshwarkar holds stock in GlaxoSmithKline, Johnson & Johnson, and Pfizer. Drs Serenko and Chan were employees of Takeda Development Center Americas at the time the study was conducted.
Dr Trivedi has received consulting fees from Abbott, Abdi Ibrahim, Akzo, Alkermes, AstraZeneca, Axon Advisors, Bristol-Myers Squibb, Cephalon, CME Institute of Physicians Postgraduate Press, Eli Lilly, Evotek, Fabre-Kramer, Forest, GlaxoSmithKline, Janssen, Johnson & Johnson, Libby, Lundbeck, Meade Johnson, MedAvante, Medtronic, Neuronetics, Otsuka, Pamlab, Parke-Davis, Pfizer, PGxHealth, Rexahn, Sepracor, Shire, Sierra, Takeda, Tai Medical/Puretech Venture, Transcept, VantagePoint, and Wyeth-Ayerst and has received research support from the Agency for Healthcare Research and Quality, Corcept Therapeutics, Cyberonics, Merck, National Alliance for Research on Schizophrenia and Depression, National Institute of Mental Health, National Institute on Drug Abuse, Naurex, Novartis, Pharmacia & Upjohn, Predix (Epix), Solvay, Targacept, and Valiant.
Funding/support: This study was supported by the Takeda Pharmaceutical Company, Ltd and H. Lundbeck A/S.
Role of the sponsor: Takeda and Lundbeck were involved in the design, investigator selection, conduct of the trial, collection of data, analysis and interpretation, and writing of the final study report. The authors had full control of the content of the manuscript.
REFERENCES
1. Ustün TB, Ayuso-Mateos JL, Chatterji S, et al. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184(5):386–392. doi:10.1192/bjp.184.5.386 PubMed
2. Angst J, Angst F, Stassen HH. Suicide risk in patients with major depressive disorder. J Clin Psychiatry. 1999;60(suppl 2):57–62, discussion 75–76,
113–116. PubMed
3. Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Survey. Arch Gen Psychiatry. 1999;56(7):617–626. doi:10.1001/archpsyc.56.7.617 PubMed
4. Trivedi MH. Major depressive disorder: remission of associated symptoms. J Clin Psychiatry. 2006;67(suppl 6):27–32. PubMed
5. Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28–40. doi:10.1176/appi.ajp.163.1.28 PubMed
6. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. doi:10.1176/ajp.2006.163.11.1905 PubMed
7. Oestergaard S, Møldrup C. Optimal duration of combined psychotherapy
and pharmacotherapy for patients with moderate and severe depression:
a meta-analysis. J Affect Disord. 2011;131(1–3):24–36. doi:10.1016/j.jad.2010.08.014 PubMed
8. Ginsberg LD. Impact of drug tolerability on the selection of antidepressant treatment in patients with major depressive disorder. CNS Spectr. 2009;14(suppl 12):8–14. PubMed
9. Ashton AK, Jamerson BD, Weinstein WL, et al. Antidepressant-related adverse effects impacting treatment compliance: Results of a patient survey. Curr Ther Res Clin Exp. 2005;66(2):96–106. doi:10.1016/j.curtheres.2005.04.006 PubMed
10. Morehouse R, Macqueen G, Kennedy SH. Barriers to achieving treatment goals: a focus on sleep disturbance and sexual dysfunction. J Affect Disord. 2011;132(suppl 1):S14–S20. doi:10.1016/j.jad.2011.03.047 PubMed
11. Bang-Andersen B, Ruhland T, Jørgensen M, et al. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder. J Med Chem. 2011;54(9):3206–3221. doi:10.1021/jm101459g PubMed
12. Westrich L, Pehrson A, Zhong H, et al. In vitro and in vivo effects for the multimodal antidepressant vortioxetine (Lu AA21004) at human and rat targets. Int J Psychiatry Clin Pract. 2012;16(suppl 1):47.
13. Sanchez C, Asin KE, Artigas F. Vortioxetine, a novel antidepressant with multimodal activity: review of preclinical and clinical data. Pharmacol Ther. 2015;145C:43–57. doi:10.1016/j.pharmthera.2014.07.001 PubMed
14. Alvarez E, Perez V, Dragheim M, et al. A double-blind, randomized, placebo-controlled, active reference study of Lu AA21004 in patients with major depressive disorder. Int J Neuropsychopharmacol. 2012;15(5):589–600. doi:10.1017/S1461145711001027 PubMed
15. Henigsberg N, Mahableshwarkar AR, Jacobsen P, et al. A randomized, double-blind, placebo-controlled 8-week trial of the efficacy and tolerability of multiple doses of Lu AA21004 in adults with major depressive disorder. J Clin Psychiatry. 2012;73(7):953–959. doi:10.4088/JCP.11m07470 PubMed
16. Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Int Clin Psychopharmacol. 2012;27(4):215–223. doi:10.1097/YIC.0b013e3283542457 PubMed
17. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138–149. doi:10.1097/YIC.0000000000000018 PubMed
18. Mahableshwarkar AR, Jacobsen PL, Chen Y, et al. NR3-055: a randomized, double-blind, duloxetine-referenced study comparing efficacy and tolerability of 2 fixed doses of vortioxetine in the acute treatment of adults with MDD. Poster presented at the 166th American Psychiatric Association (APA) Annual Meeting; May 18–22, 2013; San Francisco, CA.
19. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382–389. doi:10.1192/bjp.134.4.382 PubMed
20. American Psychiatric Association. Major depressive episode—diagnostic criteria. Diagnostic and Statistical Manual for Mental Disorders, Fourth Edition, Text Revision. Washington, DC: American Psychiatric Associaion; 2000:327.
21. Guy W. Clinical Global Impressions (028-CGI): ECDEU Assessment Manual for Psychopharmacology, Revised. Rockville, MD: US Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration; 1976:218–222.
22. Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959;32(1):50–55. doi:10.1111/j.2044-8341.1959.tb00467.x PubMed
23. Sheehan DV. The Sheehan Disability Scales. The Anxiety Disease and How to Overcome It. New York, NY: Charles Scribner & Sons; 1983:151.
24. Posner K, Brent D, Lucas C, et al. Columbia-Suicide Severity Rating Scale (C-SSRS). New York, NY: Columbia University Medical Center; 2008.
25. McGahuey CA, Gelenberg AJ, Laukes CA, et al. The Arizona Sexual Experience Scale (ASEX): reliability and validity. J Sex Marital Ther. 2000;26(1):25–40. doi:10.1080/009262300278623 PubMed
26. Rosenbaum JF, Fava M, Hoog SL, et al. Selective serotonin reuptake inhibitor discontinuation syndrome: a randomized clinical trial. Biol Psychiatry. 1998;44(2):77–87. doi:10.1016/S0006-3223(98)00126-7 PubMed
27. Naik H, Chan S, Vakilynejad M, et al. P02: a population pharmacokinetic (PK)-pharmacodynamic (PD) meta analysis of vortioxetine (Lu AA21004) in patient with major depressive disorder (MDD). Poster presented at the 166th American Psychiatric Association (APA) Annual Meeting; May 18–22, 2013; San Francisco, CA.
28. Jain R, Mahableshwarkar AR, Jacobsen PL, et al. A randomized, double-blind, placebo-controlled 6-wk trial of the efficacy and tolerability of 5 mg vortioxetine in adults with major depressive disorder. Int J Neuropsychopharmacol. 2013;16(2):313–321. doi:10.1017/S1461145712000727 PubMed
29. Baldwin DS, Loft H, Dragheim M. A randomised, double-blind, placebo controlled, duloxetine-referenced, fixed-dose study of three dosages of Lu AA21004 in acute treatment of major depressive disorder (MDD). Eur Neuropsychopharmacol. 2012;22(7):482–491. doi:10.1016/j.euroneuro.2011.11.008 PubMed
30. Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217–226. doi:10.1185/03007995.2012.761600 PubMed
31. Haddad P. Newer antidepressants and the discontinuation syndrome. J Clin Psychiatry. 1997;58(suppl 7):17–21, discussion 22. PubMed
32. Schatzberg AF, Haddad P, Kaplan EM, et al. Serotonin reuptake inhibitor discontinuation syndrome: a hypothetical definition. Discontinuation Consensus panel. J Clin Psychiatry. 1997;58(suppl 7):5–10. PubMed
33. Balon R. SSRI-associated sexual dysfunction. Am J Psychiatry. 2006;163(9):1504–1509, quiz 1664. doi:10.1176/ajp.2006.163.9.1504 PubMed
34. Mahableshwarkar AR, Jacobsen PL, Serenko M, et al. A randomized, double-blind, placebo-controlled study of the efficacy and safety of 2 doses of vortioxetine in adults with major depressive disorder. Poster presented at the 166th American Psychiatric Association (APA) Annual Meeting; May 18–22, 2013; San Francisco, CA.
This PDF is free for all visitors!