Objective: The objective of the Genomics Used to Improve DEpression Decisions (GUIDED) trial was to evaluate the utility of pharmacogenomic testing to improve outcomes among patients with major depressive disorder (MDD) who had not responded to at least 1 prior medication trial. The objective of the present analysis was to assess outcomes for the subset of patients expected to benefit from combinatorial pharmacogenomic testing because they were taking medications with predicted gene-drug interactions.
Methods: Participants (enrolled from April 14, 2014, to February 10, 2017) had an inadequate response to at least 1 psychotropic medication in the current episode of MDD. Patients were randomized to treatment as usual (TAU) or the guided-care arm, in which clinicians had access to a combinatorial pharmacogenomic test report to inform medication selection. Patients and raters were blinded to study arm through week 8. The following outcomes were assessed using the 17-item Hamilton Depre’ ‹ssion Rating Scale (HDRS-17): symptom improvement (percent change in HDRS-17 score), response (≥ 50% decrease in HDRS-17 score), and remission (HDRS-17 score ≤ 7). In the GUIDED trial, the primary endpoint of symptom improvement did not reach significance in the intent-to-treat cohort (P = .069). Here, a post hoc analysis of patients who were taking medications subject to gene-drug interactions at baseline as predicted by combinatorial pharmacogenomic testing (N = 912) is presented.
Results: Among participants taking medications subject to gene-drug interactions at baseline, outcomes at week 8 were significantly improved for those in the guided-care arm compared to TAU (symptom improvement: 27.1% versus 22.1%, P = .029; response: 27.0% versus 19.0%, P = .008; remission: 18.2% versus 10.7%, P = .003). When patients who switched medications were assessed, all outcomes were significantly improved in the guided-care arm compared to TAU (P = .011 for symptom improvement, P = .011 for response, P = .008 for remission).
Conclusions: By identifying and focusing on the patients with predicted gene-drug interactions, use of a combinatorial pharmacogenomic test significantly improved outcomes among patients with MDD who had at least 1 prior medication failure.
Trial Registration: ClinicalTrials.gov identifier: NCT02109939‘ ‹
Impact of Pharmacogenomics on Clinical Outcomes for Patients Taking Medications With Gene-Drug Interactions in a Randomized Controlled Trial

ABSTRACT
Objective: The objective of the Genomics Used to Improve DEpression Decisions (GUIDED) trial was to evaluate the utility of pharmacogenomic testing to improve outcomes among patients with major depressive disorder (MDD) who had not responded to at least 1 prior medication trial. The objective of the present analysis was to assess outcomes for the subset of patients expected to benefit from combinatorial pharmacogenomic testing because they were taking medications with predicted gene-drug interactions.
Methods: Participants (enrolled from April 14, 2014, to February 10, 2017) had an inadequate response to at least 1 psychotropic medication in the current episode of MDD. Patients were randomized to treatment as usual (TAU) or the guided-care arm, in which clinicians had access to a combinatorial pharmacogenomic test report to inform medication selection. Patients and raters were blinded to study arm through week 8. The following outcomes were assessed using the 17-item Hamilton Depression Rating Scale (HDRS-17): symptom improvement (percent change in HDRS-17 score), response (≥ 50% decrease in HDRS-17 score), and remission (HDRS-17 score ≤ 7). In the GUIDED trial, the primary endpoint of symptom improvement did not reach significance in the intent-to-treat cohort (P = .069). Here, a post hoc analysis of patients who were taking medications subject to gene-drug interactions at baseline as predicted by combinatorial pharmacogenomic testing (N = 912) is presented.
Results: Among participants taking medications subject to gene-drug interactions at baseline, outcomes at week 8 were significantly improved for those in the guided-care arm compared to TAU (symptom improvement: 27.1% versus 22.1%, P = .029; response: 27.0% versus 19.0%, P = .008; remission: 18.2% versus 10.7%, P = .003). When patients who switched medications were assessed, all outcomes were significantly improved in the guided-care arm compared to TAU (P = .011 for symptom improvement, P = .011 for response, P = .008 for remission).
Conclusions: By identifying and focusing on the patients with predicted gene-drug interactions, use of a combinatorial pharmacogenomic test significantly improved outcomes among patients with MDD who had at least 1 prior medication failure.
Trial Registration: ClinicalTrials.gov identifier: NCT02109939
J Clin Psychiatry 2019;80(6):19m12910
To cite: Thase ME, Parikh SV, Rothschild AJ, et al. Impact of pharmacogenomics on clinical outcomes for patients taking medications with gene-drug interactions in a randomized controlled trial. J Clin Psychiatry. 2019;80(6):19m12910.
To share: https://doi.org/10.4088/JCP.19m12910
© Copyright 2019 Physicians Postgraduate Press, Inc.
aPerelman School of Medicine of the University of Pennsylvania and the Corporal Michael Crescenz VAMC, Philadelphia, Pennsylvania
bUniversity of Michigan Comprehensive Depression Center and Department of Psychiatry, and National Network of Depression Centers, Ann Arbor, Michigan
cUniversity of Massachusetts Medical School and UMass Memorial Healthcare, Worcester, Massachusetts
dDepartment of Psychiatry and Behavioral Sciences, Emory University School of Medicine, Atlanta, Georgia
eDepartment of Psychiatry and Behavioral Sciences, Stanford University School of Medicine, Stanford, California
fDepartment of Psychiatry, Washington University School of Medicine, and the John Cochran Veteran’s Administration Hospital, St Louis, Missouri
gDivision of Geriatric Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Massachusetts
hDepartment of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, Maryland
iDepartment of Psychiatry and School of Medicine, University of Alabama at Birmingham, Birmingham, Alabama
jDepartment of Psychiatry and Behavioral Sciences, University of Kansas School of Medicine-Wichita, Wichita, Kansas
kAssurex Health, Inc, Mason, Ohio
lMyriad Genetics, Inc, Salt Lake City, Utah
*Corresponding author: Michael E. Thase, MD, Perelman School of Medicine of the University of Pennsylvania, The Corporal Michael Crescenz VAMC, 3535 Market Street, Room 689, Philadelphia, PA 19104 ([email protected]).
For patients with major depressive disorder (MDD), the standard treatment approach includes prescribing based on a clinician’s preference and experience as well as patients’ past treatment histories. However, too often this approach does not lead to the patient’s achieving remission—the goal of treatment in the acute phase according to the American Psychiatric Association.1 As a result, there is consensus that an improved approach to medication selection is necessary for patients with MDD. This need is especially relevant for patients with treatment-resistant depression (TRD), for whom remission rates decrease progressively as medication trials mount.2,3
Many factors may contribute to antidepressant nonresponse, including adherence, dosing, length of medication trial, and the impact of psychiatric comorbidities.4 Nonresponse also may be related to genetic alterations that adversely impact the tolerability, safety, and efficacy of psychotropic medications (ie, gene-drug interactions). As such, there has been growing interest in using pharmacogenomics to improve medication selection for those struggling with difficult-to-treat forms of MDD. For patients who are not responding to an antidepressant, pharmacogenomic testing may identify whether medication failures are influenced by gene-drug interactions. Patients and providers can then make changes in treatment regimen that may avoid or minimize the risk of gene-drug interactions at the time of testing and for future medication trials, which in turn may increase the chances of a successful outcome.
Advances in the field of pharmacogenomics have resulted in several generations of testing approaches, including single-gene testing (individual genotypes reported for single genes), multigene testing (individual genotypes reported for multiple genes), and combinatorial testing (combined phenotype reported based on algorithmic assessment of multiple genotypes). While pharmacogenomics may aid in identifying gene-drug interactions to improve prescribing, studies evaluating the utility of such tests have shown mixed results. Some of this disagreement very likely stems from differences between the different generations of pharmacogenomic testing. The limited efficacy of first-generation pharmacogenomic testing of individual genes that encode for cytochrome P450 (CYP) has now been well established.5 This limited efficacy is consistent with our growing understanding of the complexity of drug metabolism. When second-generation tests incorporated more genes, mixed results were still reported for multigene pharmacogenomic testing, with some evidence of utility for patients with severe depression.6
The third generation of pharmacogenomic testing in MDD accounts for the combined effect of multiple genotypes that may impact the pharmacokinetics or pharmacodynamics of a drug. This combinatorial approach to pharmacogenomic testing has demonstrated clinical validity and utility in medication selection among patients with TRD7-10; however, concerns about study design (ie, cohort size and blinding) have called these findings into question.11-14
To address many of these concerns, a recent large, blinded, randomized controlled trial (Genomics Used to Improve DEpression Decisions [GUIDED])15 was conducted. GUIDED evaluated the utility of using combinatorial pharmacogenomic testing to inform medication selection (guided care) compared to standard prescribing approaches (treatment as usual) for patients with MDD and at least 1 failed medication trial.15 The primary outcome of symptom improvement was not significantly different between study arms in the intent-to-treat cohort (P = .069). However, the rates of response and remission were significantly improved in the guided-care arm compared to treatment as usual,15 which suggests that a subgroup of patients did obtain a clinically meaningful benefit from pharmacogenomic testing. This benefit may be related to whether medication failures were due to gene-drug interactions, as significant improvements in all evaluated outcomes were observed among patients who entered the study taking medications with significant gene-drug interactions.
It is assumed that pharmacogenomic testing would be of greatest potential benefit to patients whose nonresponse is mediated by gene-drug interactions. Although patients must be tested to identify gene-drug interactions, those who are not taking medications with gene-drug interactions can dilute the measured efficacy of using the pharmacogenomic testing to inform medication selection in a trial. This was the case in the GUIDED trial, in which approximately 30% of patients entered the study taking medications with no predicted gene-drug interactions.
To more directly evaluate the utility of pharmacogenomic testing in informing treatment decisions among patients with TRD, we sought to evaluate the extent to which pharmacogenomic testing positively influenced antidepressant outcomes in those patients entering the GUIDED trial taking medications subject to gene-drug interactions. Patient outcomes were compared for all patients in this subset as well as for those who switched medications after baseline.
METHODS
Participants and Study Design
The GUIDED trial was a 24-week randomized controlled trial to evaluate outcomes among patients with MDD when combinatorial pharmacogenomic testing was used to guide medication selection compared to treatment as usual (TAU). The trial was approved by the Copernicus Group independent review board (INC1-14-012) and has been previously described in detail.15 In brief, patients over the age of 18 years were eligible if they were diagnosed with MDD according to both the self-rated and site-rated 16-item Quick Inventory of Depression Symptomatology (QIDS-SR-16 and QIDS-C-16 score ≥ 11)16 at screening and baseline and had at least 1 failed medication trial (inadequate therapeutic response or intolerable side effects) within the current depressive episode. Written informed consent was obtained, and the study was registered at ClinicalTrials.gov (identifier: NCT02109939).
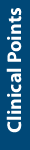
- An improved approach to medication selection is necessary for patients with major depressive disorder (MDD), especially for those with treatment-resistant depression.
- For patients with MDD who were taking medications with gene-drug interactions at baseline, having access to combinatorial pharmacogenomic test results to inform medication selection resulted in significantly improved patient outcomes compared to treatment as usual.
Patients (enrolled from April 14, 2014, to February 10, 2017) were randomized 1:1 to TAU or the guided-care arm, in which combinatorial pharmacogenomic test results were available to guide treatment decisions. Testing was performed prior to the baseline visit so that the report would be available at the baseline visit for patients in the guided-care arm. Providers were not required to take action in response to test results.
Patients and raters in both arms were blinded to study arm and test results. To incorporate the pharmacogenomic test report in prescribing, providers could not be blinded to study arm; however, they were blinded to test results for patients in TAU. All blinding was maintained through week 8. Sites were instructed to unblind after week-12 assessments, though unblinding may have occurred before assessments were performed. As a result, data collected through week 8 were considered blinded.
Outcomes were assessed at baseline, week 4, week 8, week 12, and week 24. The 17-item Hamilton Depression Rating Scale (HDRS-17), which was the primary assessment of outcome, was administered by central raters (MedAvante-ProPhase Inc; Hamilton Township, New Jersey) without knowledge of treatment arm.
Combinatorial PGx testing. All patients were tested with the GeneSight Psychotropic test (Assurex Health Inc; Mason, Ohio). The test has been previously described in detail.17 In brief, the genotypes of 59 alleles and variants across 8 genes were evaluated (CYP1A2: −3860G > A, −2467T > delT, −739T > G, −729C > T, −163C > A, 125C > G, 558C > A, 2116G > A, 2473G > A, 2499A > T, 3497G > A, 3533G > A, 5090C > T, 5166G > A, 5347C > T; CYP2C9: *1, *2, *3, *4, *5, *6; CYP2C19: *1, *2, *3, *4, *5, *6, *7, *8, *17; CYP3A4: *1, *13, *15A, *22; CYP2B6: *1, *4, *6, *9; CYP2D6: *1, *2, *2A, *3, *4, *5 (gene deletion), *6, *7, *8, *9, *10, *11, *12, *14, *15, *17, *41, gene duplication; HTR2A: −1438 G > A; and SLC6A4: L, S).
The individual genotype for each variant or allele was included in a weighted algorithmic assessment for each individual medication. The algorithmic assessment for a medication included all tested genes implicated in the pharmacokinetics or pharmacodynamics for that medication. A combined phenotype was produced for each medication to account for the combined effect of all genetic alterations in relevant genes. These combined phenotypes were used to categorize 38 psychotropic medications according to the predicted level of gene-drug interactions. The report categories were “use as directed” (no gene-drug interactions), “use with caution” (moderate gene-drug interactions), and “use with increased caution and with more frequent monitoring” (significant gene-drug interactions). Depending on the type and severity of predicted gene-drug interactions, recommendations may have included dose modification, consideration of increased side effect risk, or consideration of reduced efficacy or that the medication is contraindicated.
Statistical Analysis
This post hoc analysis focused on the participants who were taking medications subject to gene-drug interactions at baseline (“use with caution” and “use with increased caution and with more frequent monitoring” report categories). This subset was drawn from the intent-to-treat (ITT) study population, which included all patients who were randomized and met inclusion and exclusion criteria at baseline. Patient outcomes at the blinded week-8 endpoint were compared and included symptom improvement (percent change in HDRS-17 score from baseline), response (≥ 50% decrease in HDRS-17 score), and remission (HDRS-17 score ≤ 7). The longer-term value of pharmacogenomic testing was evaluated via patient outcomes through the full 24-week study period. As TAU ended after week 8 when the pharmacogenomic test report was made available, this group was not included in the analyses of longer-term outcomes.
Medication switches were defined as stopping at least 1 medication and adding at least 1 different medication. The proportion of patients who made medication switches from baseline to week 8 were compared for the 2 study arms. Patient outcomes at week 8 also were evaluated among the subset of patients who switched medications.
The percentage change from baseline to week 8 in HDRS-17 score was conducted by fitting a mixed model for repeated measures. The percentage of responders and remitters as determined via HDRS-17 score was analyzed separately by fitting a generalized linear mixed model. Both models included treatment, week, treatment-by-week interaction, baseline HDRS-17 score, and baseline HDRS-17 score-by-week interaction as fixed effects. The primary comparison between the 2 treatment arms at week 8 was tested at a significance level of .05 (2-sided).
RESULTS
Cohort
A total of 1,799 patients were randomized to guided care (n = 899) or TAU (n = 900) in GUIDED, of whom 1,541 patients completed the baseline visit and composed the ITT sample (760 in the guided-care arm, 781 in TAU).15 A total of 912 patients (59%) were taking medications with predicted gene-drug interactions at baseline (439 in the guided-care arm, 473 in TAU; Supplementary Figure 1) and were included in this analysis. There were no substantial differences in demographics or disease severity for patients who were included for analysis and those who were not taking any baseline medications with gene-drug interactions (Supplementary Table 1).
Click figure to enlarge
Click figure to enlarge
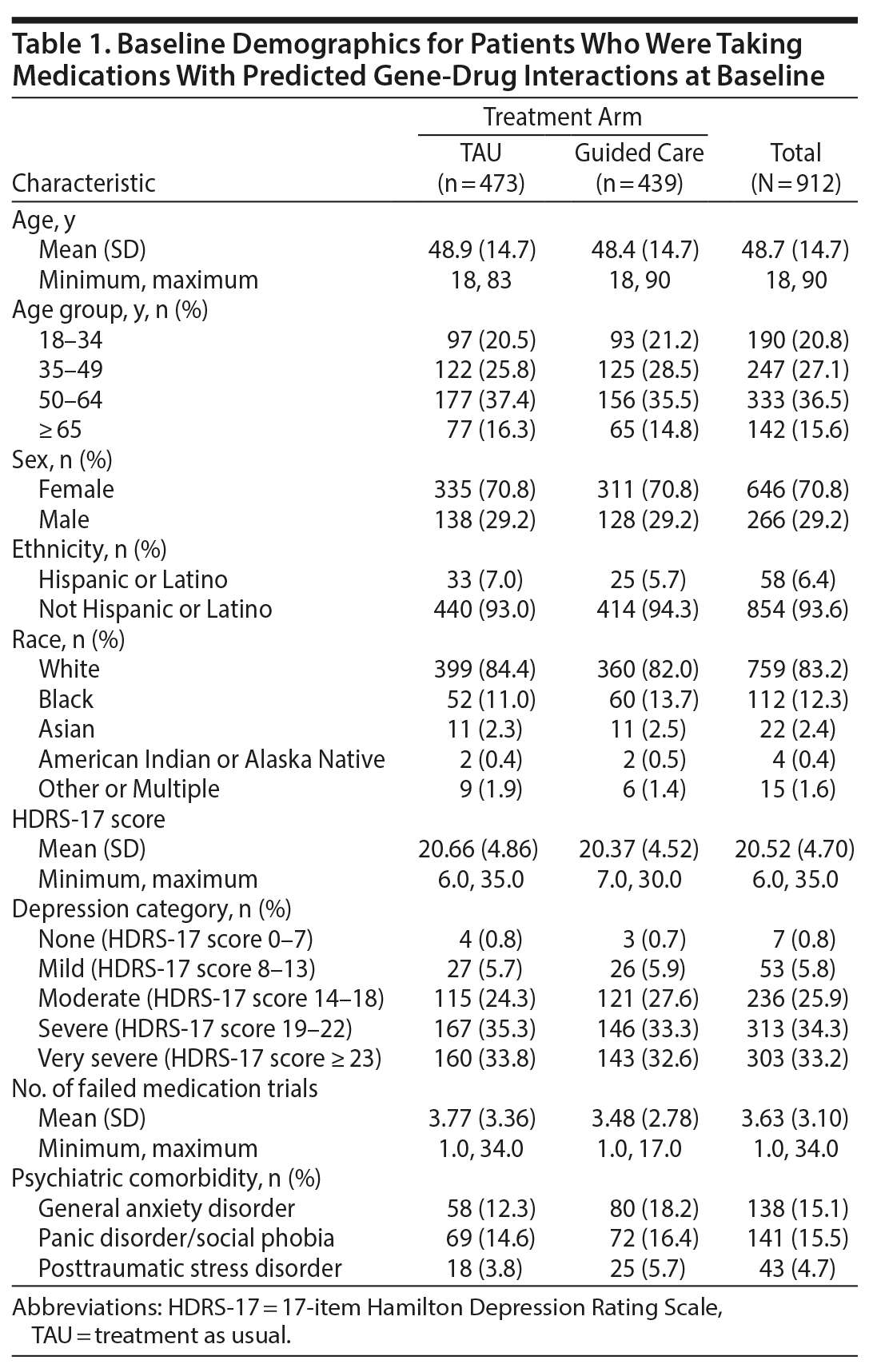
Among patients who were taking medications with gene-drug interactions at baseline, the mean age at testing was 48.7 years (Table 1). The majority of patients were female (70.8%) and non-Hispanic/Latino (93.6%). The mean number of failed medications was 3.6 (Table 1). The mean HDRS-17 score was 20.5, with MDD severity ranging from mild to very severe (Table 1). There was a small number of patients with minimal depression at baseline according to HDRS-17 score, reflecting the fact that inclusion criteria were based on site- or self-rated QIDS-16 rather than the central-rated HDRS-17. For patients taking medications subject to gene-drug interactions at baseline, there were no substantial differences in demographics or disease between arms at baseline (Table 1).
In the guided-care arm, 82 patients were lost to follow-up or discontinued the study prior to week 8. For TAU, only 43 patients were lost to follow-up or discontinued the study. A total of 787 patients taking medications with predicted gene-drug interactions at baseline completed the study through week 8 (357 in the guided-care arm, 430 in TAU; Supplementary Figure 1).
Patient Outcomes at Week 8
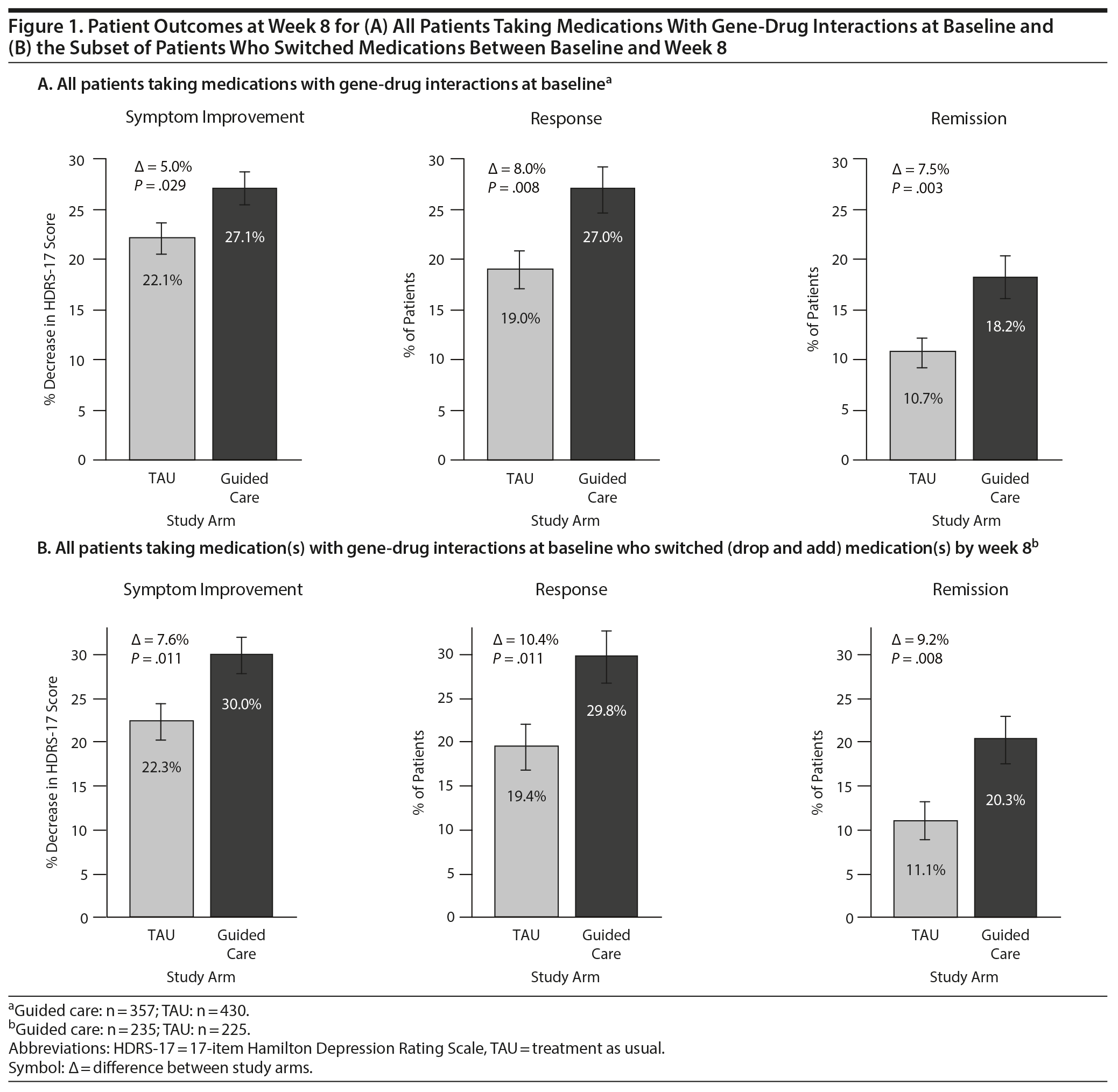
Among patients taking medications with predicted gene-drug interactions at baseline, HDRS-17 scores decreased by 27.1% from baseline to week 8 in the guided-care arm compared to 22.1% in TAU (Figure 1A). This represented a significant difference in symptom improvement in the guided-care arm compared to TAU (−† = 5.0%, P = .029). At week 8, the response rate was 27.0% in the guided-care arm compared to 19.0% in TAU (−† = 8.0%, P = .008; Figure 1A). At week 8, the remission rate was 18.2% in the guided-care arm compared to 10.7% in TAU (−† = 7.5%, P = .003; Figure 1A).
Durability of Guided-Care Outcomes Through Week 24
Patient outcomes in the guided-care arm continued to improve through week 24 (Figure 2). There was a 42.2% decrease in HDRS-17 scores at week 24 compared to baseline. This decrease in HDRS-17 score represents a 56% increase in symptom improvement from week 8. At week 24, the response rate was 44.3% and the remission rate was 33.2%. These response rates represent a 64% and an 82% improvement, respectively, from week 8.
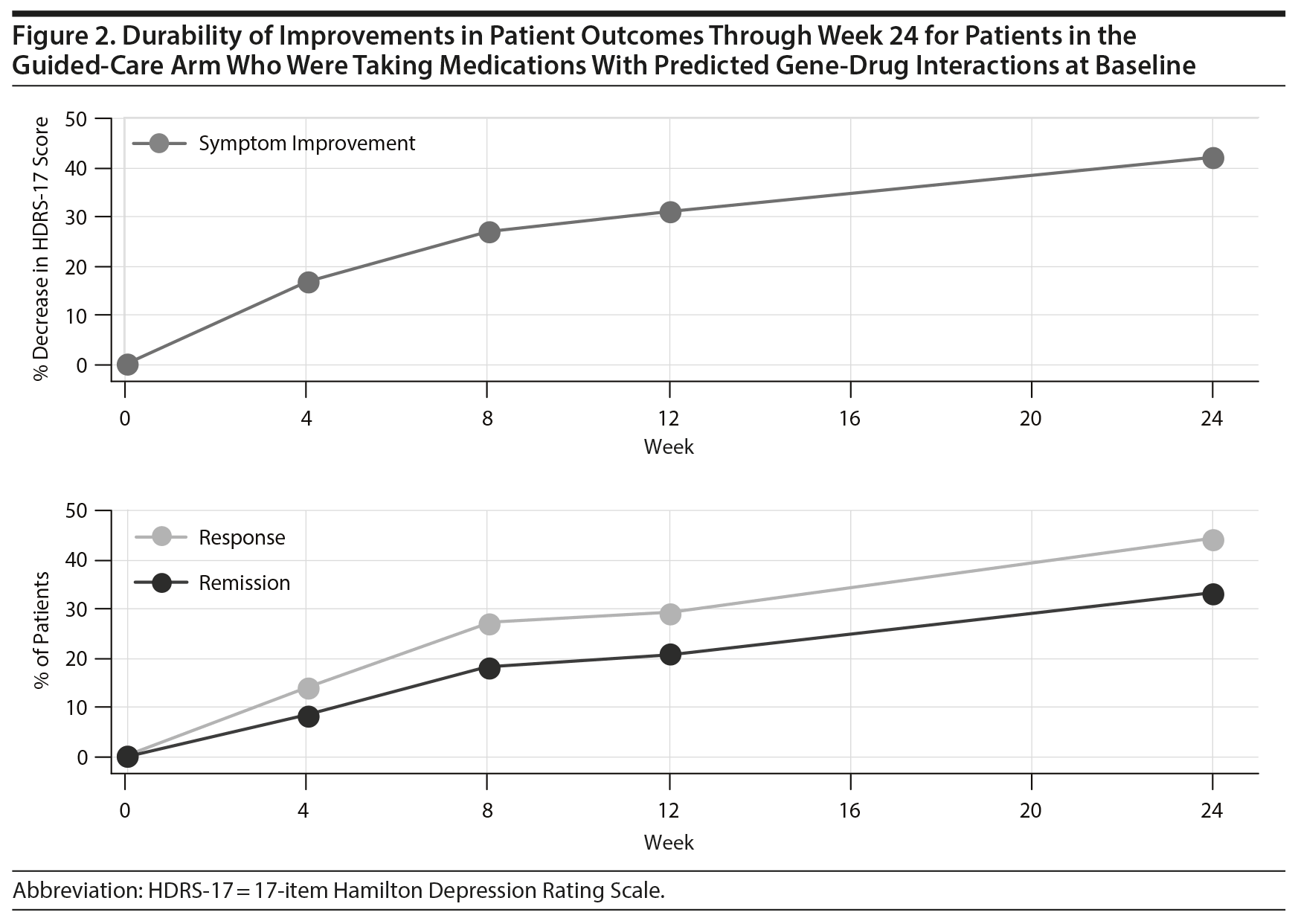
Click figure to enlarge
Patient Outcomes After Medication Switches
Medication switches (dropping a medication and adding a different medication) during the first 8 weeks of treatment were significantly more common in the guided-care arm (65.8%; 235/357) than in the TAU arm (52.3%; 225/430) (P < .001). Among patients who switched medications, HDRS-17 scores decreased by 30.0% from baseline to week 8 in the guided-care arm compared to 22.3% in TAU (−† = 7.6%, P = .011; Figure 1B).
Among patients who switched medications, the rates of response and remission were significantly improved for those in the guided-care arm compared to TAU. The response rate at week 8 among patients who switched medications was 29.8% for those in the guided-care arm compared to 19.4% for TAU (−† = 10.4%, P = .011; Figure 1B). Similarly, the remission rate at week 8 for patients who switched medications in the guided-care arm was 20.3% compared to 11.1% in TAU (−† = 9.2%, P = .008; Figure 1B). This represents a relative improvement of 54% for response and 83% for remission among patients whose medication switches were informed by pharmacogenomic testing.
The improved patient outcomes observed for the guided-care arm compared to TAU reflect the types of medication switches made in each arm. Among those who switched medications, the proportions of patients taking baseline medications with moderate or significant gene-drug interactions were comparable (Figure 3). In comparison, 66.4% of patients in the guided-care arm switched to medications with no gene-drug interactions by week 8, while only 20.0% of patients in TAU switched to medications with no gene-drug interactions (Figure 3). Of note, these changes in TAU were not informed by the patients’ pharmacogenomic test results as providers were blinded to those results until after week 8.
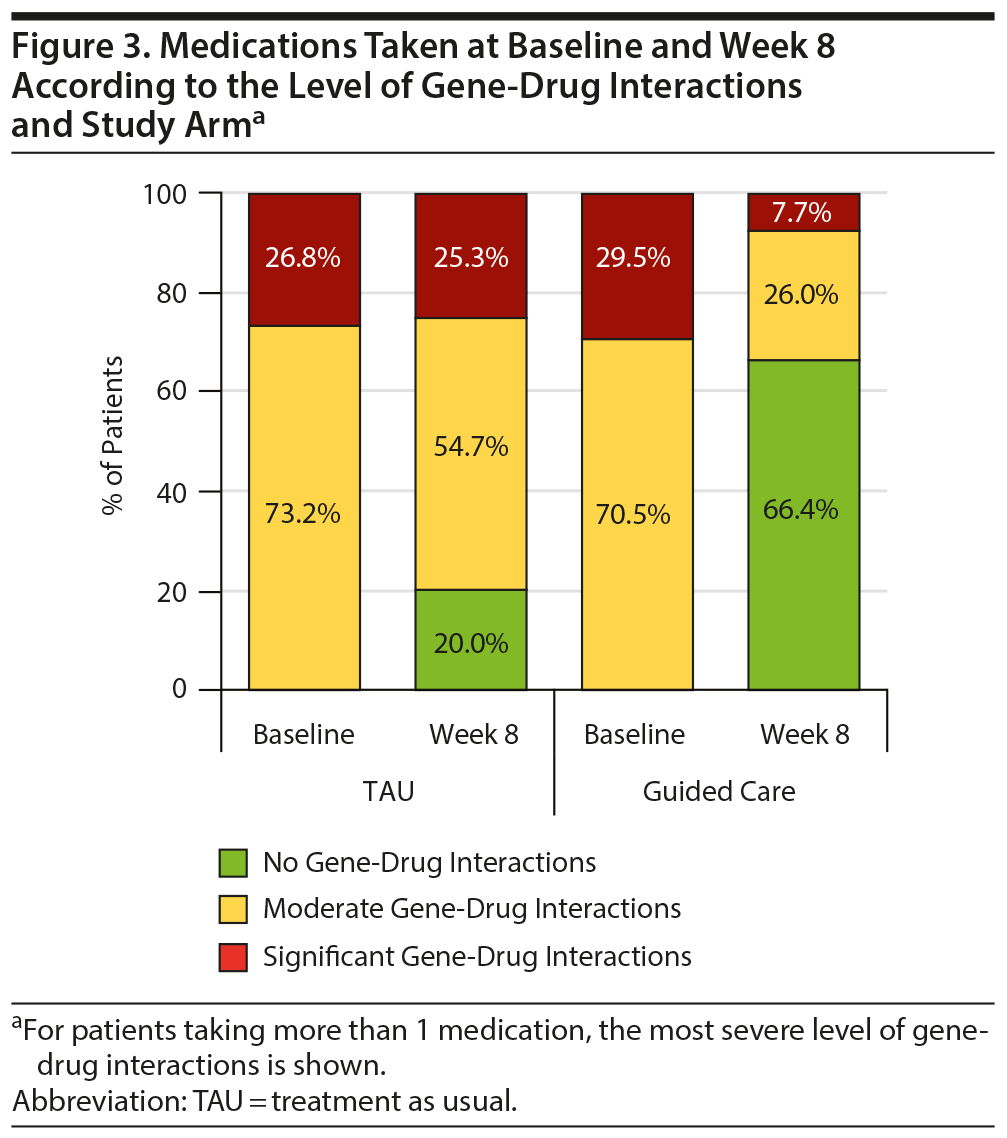
Click figure to enlarge
DISCUSSION
Pharmacogenomic testing has been explored as a precision-treatment strategy to improve medication selection in patients with MDD. With different available testing approaches, the collective evidence regarding the utility of pharmacogenomic testing in MDD has been mixed. This notion is consistent with a recent study demonstrating that different pharmacogenomic tests and testing approaches are not equivalent and must be evaluated separately.4 A recent large randomized controlled trial (GUIDED)15 demonstrated that utilization of a combinatorial pharmacogenomic test to inform medication selection resulted in improved response and remission among patients with TRD. However, that study did not achieve significance in the primary objective—symptom improvement. Because pharmacogenomic testing has the most potential to help patients who are taking medications affected by gene-drug interactions, improvements in patient outcomes in GUIDED were diluted by patients taking baseline medications with no gene-drug interactions.
To more directly evaluate the impact of pharmacogenomic testing in this subanalysis, we evaluated patients from the GUIDED trial who were taking medications predicted to have gene-drug interactions at baseline. Among these patients, symptom improvement, response rate, and remission rate were all significantly improved in the guided-care arm compared to TAU. This finding demonstrates the utility of pharmacogenomic testing in guiding medication selection for patients who are likely failing a medication for genetic reasons. Although patients taking medications with gene-drug interactions can be identified only after pharmacogenomic testing, previous work has demonstrated that combinatorial pharmacogenomic testing for patients with TRD is cost-effective.18
The low rates of response and remission reported for TAU are consistent with published reports of well-controlled TRD studies2,19-21 and highlight the clinical challenge of treating TRD. Despite the fact that TAU patients received active treatment prescribed according to standard practice, only 10.7% reached remission at week 8. When combinatorial pharmacogenomic testing was available to inform medication selection, remission rates improved by nearly 60% (18.2%). The rate of remission was still modest due to the degree of treatment resistance and medication failures for non-genetic reasons; however, this finding represents a clinically important improvement for this challenging to treat population.
Patient outcomes in the guided-care arm were also evaluated over the full 24-week study period to assess the durability of patient outcomes when pharmacogenomic testing was used to inform medication selection. All patient outcomes in the guided-care arm were durable through the full 24-week study. In fact, outcomes in the guided-care arm continued to improve: the rate of remission nearly doubled from week 8 to week 24. This observation supports that pharmacogenomic testing may provide durability in antidepressant effects and aid in sustaining antidepressant improvements in the maintenance therapy setting.
Pharmacogenomic testing can be pivotal in identifying when genetic factors contribute to medication failures, allowing providers to make data-driven decisions to change a patient’s treatment regimen while also informing the selection of new medications to avoid additional gene-drug interactions. Changes in treatment regimen were not mandated as part of the GUIDED trial. To this end, a larger proportion of patients in the guided-care arm switched medications between baseline and week 8 compared to TAU. To determine whether improved patient outcomes were due to medication switches in general or pharmacogenomic-guided switches, we evaluated the utility of pharmacogenomic testing in the subset of patients who switched medications between baseline and week 8 in both arms. This subanalysis showed that all patient outcomes were significantly improved among patients whose medication switches were informed by pharmacogenomic testing compared to those whose medication switches were not informed by pharmacogenomics. While some studies22,23 have suggested that medication changes in general may improve patient outcomes, the data from the present study demonstrated that patient outcomes were significantly better when pharmacogenomic test results were available to inform changes in treatment.
There were some limitations of this analysis. First, many non-genetic factors may contribute to medication failure. These factors were not explicitly collected in the GUIDED trial, and thus their impact on patient outcomes cannot be assessed. However, any impact of non-genetic factors should affect both study arms equally due to balanced randomization. In addition, we were unable to address the utility of pharmacogenomic testing to inform multiple medication trials against TAU. This limitation may be relevant for patients who changed to medications with no gene-drug interactions that failed for non-genetic reasons. In these cases, the availability of pharmacogenomic testing may result in improved patient outcomes over a longer interval than was evaluated in the blinded study period. However, this long-term utility is supported by the increase in the proportion of patients who achieved response and remission in the guided-care arm from week 8 to week 24; these results very likely include multiple medication trials for a subset of patients.
In summary, treatment decisions supported by pharmacogenomic testing resulted in improved symptom improvement, response, and remission among patients enrolled in a large randomized controlled trial who entered the study taking medications with gene-drug interactions. All patient outcomes remained significantly improved for the guided-care arm versus TAU when the subset of patients who switched medications were evaluated separately. Patients with treatment-resistant depression have a low likelihood of reaching remission with standard treatment approaches. Collectively, the data presented here support the utility of pharmacogenomic testing in patients who are failing their current medications due to genetic reasons. Identifying these gene-drug interactions can prompt appropriate changes in prescribing to ultimately improve patient outcomes.
Submitted: May 13, 2019; accepted October 10, 2019.
Published online: October 31, 2019.
Potential conflicts of interest: Dr Thase has received research support from Assurex Health, Acadia, Agency for Healthcare Research and Quality, Alkermes, Avanir, Forest, Intracellular, Janssen, National Institute of Mental Health (NIMH), Otsuka, Patient-Centered Outcomes Research Institute, and Takeda; has served as a consultant for Acadia, Akili, Alkermes, Allergan (Forest, Naurex), AstraZeneca, Cerecor, Eli Lilly, Fabre-Kramer, Gerson Lehrman Group, Guidepoint Global, Johnson & Johnson (Janssen, Ortho-McNeil), Lundbeck, MedAvante, Merck, Moksha8, Nestlé (PamLab), Novartis, Otsuka, Pfizer, Shire, Sunovion, and Takeda; and receives royalties from American Psychiatric Press, Guilford Publications, Herald House, and W.W. Norton & Company, Inc. Dr Parikh has received research funding from the Ontario Brain Institute, the Canadian Institutes of Health Research, and the James and Ethel Flinn Foundation; has served as a consultant for Assurex Health; has received honoraria from Mensante Corporation, Takeda, and the Canadian Network for Mood and Anxiety Treatments (CANMAT); and has equity in Mensante. Dr Rothschild has received research support from Allergan, AssureRx, Janssen, NIMH, Takeda, Eli Lilly, and Pfizer; has served as a consultant for Alkermes, Eli Lilly, GlaxoSmithKline, Myriad Genetics, Pfizer, SageTherapeutics, and Sanofi-Aventis; and receives royalties for the Rothschild Scale for Antidepressant Tachyphylaxis (RSAT); Clinical Manual for the Diagnosis and Treatment of Psychotic Depression, American Psychiatric Press, 2009; The Evidence-Based Guide to Antipsychotic Medications, American Psychiatric Press, 2010; The Evidence-Based Guide to Antidepressant Medications, American Psychiatric Press, 2012; and UpToDate (Wolters Kluwer). Dr Dunlop has received research support from Acadia, Assurex Health, Axsome, Janssen, and Takeda and has served has a consultant for Assurex Health and Aptinyx. Dr DeBattista has received research support from Assurex Health and Brain Resources. Dr Conway has received research support from LivaNova, Bristol-Myers Squibb, the Stanley Medical Research Institute, NIMH, NeoSync Inc, The Taylor Family Institute for Innovative Psychiatric Research, The August Busch IV Foundation, and the Barnes-Jewish Hospital Foundation; has received speaking fees from Bristol-Myers Squibb and Otsuka; has served as a research design consultant for LivaNova; and is a part-time employee of the John Cochran Veterans Administration Hospital in St. Louis, Missouri. Dr Forester has received research funding from the National Institutes of Health, Rogers Family Foundation, Spier Family Foundation, Assurex Health, Eli Lilly, and Biogen and has served as a consultant for Biogen. Dr Mondimore has received research funding from Assurex Health. Dr Shelton has received research funding from Acadia, Alkermes, Allergan, Assurex Health, Avanir, Cerecor, Genomind, Intracellular Therapies, Janssen, Otsuka, and Takeda and has served as a consultant for Acadia, Allergan, Cerecor, Janssen, Lundbeck, and Takeda. Dr Macaluso has conducted clinical trials research as principal investigator for Acadia, Alkermes, Allergan, Assurex Health, Eisai, Lundbeck, Janssen, Naurex/Aptinyx, and Neurim; all study contracts and payments were made to Kansas University Medical Cancer Research Institute. Drs Li and Jablonski are employed by Assurex Health (now Myriad Neuroscience). Dr Brown is employed by Myriad Genetics Inc. Dr Greden has served as a scientific advisor for Janssen, Naurex (Allergan), Cerecor, NeuralStem, Sage Therapeutics, and Genomind and received reimbursement as a speaker for Assurex Health in 2014; all work was done as an unpaid consultant to Assurex and Myriad.
Funding/support: This study was supported by Assurex Health (now Myriad Neuroscience, Mason, Ohio). Assurex Health provided testing in kind.
Role of the sponsor: Assurex Health (now Myriad Neuroscience, Mason, Ohio) provided testing and participated in the study design for the original trial. Authors who are employed by the sponsor participated in data analysis, data interpretation, and manuscript drafting as part of their roles as authors.
Supplementary material: See accompanying pages.
REFERENCES
1.Armstrong C. APA releases guideline on treatment of patients with major depressive disorder. Am Fam Physician. 2011;83(10):1219-1227.
2.Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917. PubMed CrossRef
3.Greden JF, Riba MB, McInnis MG, eds. Treatment Resistant Depression: A Roadmap for Effective Care. Washington, DC: American Psychiatric Publishing, Inc.; 2011.
4.Bousman CA, Dunlop BW. Genotype, phenotype, and medication recommendation agreement among commercial pharmacogenetic-based decision support tools. Pharmacogenomics J. 2018;18(5):613-622. PubMed CrossRef
5.Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: testing for cytochrome P450 polymorphisms in adults with nonpsychotic depression treated with selective serotonin reuptake inhibitors. Genet Med. 2007;9(12):819-825. PubMed CrossRef
6.Bradley P, Shiekh M, Mehra V, et al. Improved efficacy with targeted pharmacogenetic-guided treatment of patients with depression and anxiety: a randomized clinical trial demonstrating clinical utility. J Psychiatr Res. 2018;96:100-107. PubMed CrossRef
7.Altar CA, Carhart JM, Allen JD, et al. Clinical validity: Combinatorial pharmacogenomics predicts antidepressant responses and healthcare utilizations better than single gene phenotypes. Pharmacogenomics J. 2015;15(5):443-451. PubMed CrossRef
8.Hall-Flavin DK, Winner JG, Allen JD, et al. Utility of integrated pharmacogenomic testing to support the treatment of major depressive disorder in a psychiatric outpatient setting. Pharmacogenet Genomics. 2013;23(10):535-548. PubMed CrossRef
9.Hall-Flavin DK, Winner JG, Allen JD, et al. Using a pharmacogenomic algorithm to guide the treatment of depression. Transl Psychiatry. 2012;2(10):e172. PubMed CrossRef
10.Winner JG, Carhart JM, Altar CA, et al. A prospective, randomized, double-blind study assessing the clinical impact of integrated pharmacogenomic testing for major depressive disorder. Discov Med. 2013;16(89):219-227. PubMed
11.Goldberg JF. Do you order pharmacogenetic testing? why? J Clin Psychiatry. 2017;78(8):1155-1156. PubMed CrossRef
12.Rosenblat JD, Lee Y, McIntyre RS. Does pharmacogenomic testing improve clinical outcomes for major depressive disorder? a systematic review of clinical trials and cost-effectiveness studies. J Clin Psychiatry. 2017;78(6):720-729. PubMed CrossRef
13.Zeier Z, Carpenter LL, Kalin NH, et al. Clinical implementation of pharmacogenetic decision support tools for antidepressant drug prescribing. Am J Psychiatry. 2018;175(9):873-886. PubMed CrossRef
14.Zubenko GS, Sommer BR, Cohen BM. On the marketing and use of pharmacogenetic tests for psychiatric treatment. JAMA Psychiatry. 2018;75(8):769-770. PubMed CrossRef
15.Greden JF, Parikh SV, Rothschild AJ, et al. Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the GUIDED trial: a large, patient- and rater-blinded, randomized, controlled study. J Psychiatr Res. 2019;111:59-67. PubMed CrossRef
16.Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), Clinician Rating (QIDS-C), and Self-Report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry. 2003;54(5):573-583. PubMed CrossRef
17.Jablonski MR, King N, Wang Y, et al. Analytical validation of a psychiatric pharmacogenomic test. Per Med. 2018;15(3):189-197. PubMed CrossRef
18.Winner JG, Carhart JM, Altar CA, et al. Combinatorial pharmacogenomic guidance for psychiatric medications reduces overall pharmacy costs in a 1 year prospective evaluation. Curr Med Res Opin. 2015;31(9):1633-1643. PubMed CrossRef
19.Lam RW, Chan P, Wilkins-Ho M, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and metaanalysis. Can J Psychiatry. 2008;53(9):621-631. PubMed CrossRef
20.Mahmoud RA, Pandina GJ, Turkoz I, et al. Risperidone for treatment-refractory major depressive disorder: a randomized trial. Ann Intern Med. 2007;147(9):593-602. PubMed CrossRef
21.McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry. 2006;163(9):1531-1541, quiz 1666. PubMed CrossRef
22.Thase ME, Rush AJ, Howland RH, et al. Double-blind switch study of imipramine or sertraline treatment of antidepressant-resistant chronic depression. Arch Gen Psychiatry. 2002;59(3):233-239. PubMed CrossRef
23.Papakostas GI, Fava M, Thase ME. Treatment of SSRI-resistant depression: a meta-analysis comparing within- versus across-class switches. Biol Psychiatry. 2008;63(7):699-704. PubMed CrossRef
This PDF is free for all visitors!