ABSTRACT
Objective: To describe the tolerability of esketamine nasal spray based on the adverse event profile observed during treatment sessions occurring early and later over the course of treatment.
Methods: In 2 long-term, phase 3 studies (NCT02493868, October 1, 2015–February 16, 2018; NCT02497287, September 30, 2015–October 28, 2017), patients with treatment-resistant major depressive disorder (per DSM-5) and nonresponse to ≥ 2 oral antidepressants received esketamine nasal spray (56 or 84 mg) twice weekly during a 4-week induction phase, weekly for weeks 5–8, and weekly or every 2 weeks thereafter as maintenance treatment, in conjunction with a new oral antidepressant. A post hoc analysis using descriptive statistics evaluated occurrence (incidence, frequency, severity) and recurrence (incidence and severity) of events of specific interest.
Results: In patients treated with esketamine nasal spray plus a newly initiated oral antidepressant (n = 928), spontaneously reported adverse events of dizziness, nausea, sedation, vertigo, and increased blood pressure were more likely to recur after the first week of treatment if they occurred more frequently (twice > once > none) during the first week. The same pattern was observed when these events were assessed by structured instruments. Incidences of dizziness, dissociation, increased blood pressure, nausea, vertigo, and sedation were highest in week 1 of treatment (20.6%, 16.7%, 4.3%, 14.0%, 12.1%, and 3.8%, respectively) and decreased thereafter. Initial occurrences and subsequent recurrences of events were mostly mild or moderate in severity.
Conclusions: Adverse events during treatment with esketamine nasal spray plus an oral antidepressant generally become less frequent with ongoing treatment, and the majority are mild or moderate in severity.
Trial Registration: ClinicalTrials.gov identifiers: NCT02493868; NCT02497287
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
J Clin Psychiatry 2022;83(6):21m14318
To cite: Williamson DJ, Gogate JP, Kern Sliwa JK, et al. Longitudinal course of adverse events with esketamine nasal spray: a post hoc analysis of pooled data from phase 3 trials in patients with treatment-resistant depression. J Clin Psychiatry. 2022;83(6):21m14318.
To share: https://doi.org/10.4088/JCP.21m14318
© 2022 Physicians Postgraduate Press, Inc.
aField-Based Medical Affairs, Janssen Scientific Affairs, LLC, Titusville, New Jersey
bStatistics and Decision Sciences, Janssen Research & Development, LLC, Titusville, New Jersey
cNeuroscience Medical Information, Janssen Scientific Affairs, LLC, Titusville, New Jersey
dJanssen Medical Information, Janssen Scientific Affairs, LLC, Titusville, New Jersey
eDepartment of Psychiatry and Behavioral Sciences, University of Kansas School of Medicine, Wichita, Kansas
fDepartment of Psychiatry, UConn Health, Farmington, Connecticut
gClinical Development, Janssen Scientific Affairs, LLC, Titusville, New Jersey
‡Drs Williamson and Daly and Mr Manera were affiliated with Janssen Scientific Affairs, LLC, at the time the trial was conducted.
*Corresponding author: David J. Williamson, PhD, University of South Alabama College of Medicine, 2450 Old Shell Rd, Ste A, Mobile, AL 36607 ([email protected]).
Medication management for an individual patient is optimized by balancing the potential for benefit versus harm. Patients are not equally susceptible to every adverse event (AE) listed in a product’s prescribing information; rates provided therein generally do not provide information about how the rate and severity of specific AEs change over time with repeated dosing.
Esketamine nasal spray (ESK) is a noncompetitive N-methyl-d-aspartate receptor antagonist approved, in conjunction with an oral antidepressant (OAD), for the treatment of adults with treatment-resistant major depressive disorder (TRD) or major depressive disorder with acute suicidal ideation or behavior.1–8 The US Food and Drug Administration (FDA) requires a Risk Evaluation and Mitigation Strategy (REMS) for ESK to ensure its benefits outweigh the risks of misuse, abuse, and serious adverse outcomes from sedation and dissociation, which includes a requirement of post-administration monitoring for ≥ 2 hours.9 Two notable characteristics of the ESK treatment paradigm are discrete treatment sessions lasting ≥ 2 hours and treatment-associated AEs that are transient and generally resolve within 90 minutes.6,10–13 A pooled analysis of patients who participated in the TRANSFORM-1 (NCT02417064) and TRANSFORM-2 (NCT02418585) studies, two 4-week, phase 3, double-blind, placebo-controlled, multicenter studies of ESK in patients with treatment-resistant depression, found that across treatment sessions, ≥ 90% of patients were considered ready for discharge at 90 minutes after administration, with an additional 2%–7% ready at 2 hours.11 The ≥ 2-hour ESK treatment sessions and transient AEs associated with ESK treatment provide a valuable opportunity to determine how a patient’s early experience of AEs may impact the likelihood of recurrence with subsequent administrations. In addition to assessing the patterns of AE occurrence over time following repeated administrations of ESK, we investigated the following questions:
- Does the frequency of AEs observed during the initial weeks of treatment (induction) provide any indication of the rates of AEs observed later in treatment?
- Does the severity of AEs reported during the initial weeks of treatment provide any indication of the reported severity of AE recurrences, should they occur?
- Do the incidence and severity of AEs occurring during the last week of induction treatment (week 4) provide a better indication of recurrence and/or severity with longer-term treatment than those observed during week 1?
- To the extent that later AE frequency or severity differ according to what is observed early in treatment, does ESK dose or medications given to prevent or attenuate these AEs appear to affect an observed pattern?
METHODS
Patients
Data were pooled from adults (aged 18–64 years) with TRD enrolled in the SUSTAIN-11 (NCT02493868, October 1, 2015–February 16, 2018) and SUSTAIN-27 (NCT02497287, September 30, 2015–October 28, 2017) studies. Patients had recurrent or single-episode (≥ 2 years) major depressive disorder (Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition) and met study-defined criteria for TRD. In the relapse prevention study, SUSTAIN-1, at screening, depressive symptoms had been nonresponsive to adequate trials of ≥ 1 and ≤ 5 OADs in the current depressive episode.1 Nonresponse to a different, ongoing OAD was confirmed at the end of the 4-week screening prospective observational phase (defined as ≤ 25% improvement in Montgomery-Asberg Depression Rating Scale [MADRS] total score from week 1 to week 4 and a MADRS total score of ≥ 28 on week 2 and week 4).1 In contrast, in the open-label safety study, SUSTAIN-2, patients must have had ≥ 2 prior OAD failures in the current episode (determined retrospectively) and a MADRS total score of ≥ 22.7
Study Designs
The SUSTAIN studies, and the 2 short-term trials (TRANSFORM-1 and -2 in adults aged < 65 years) from which patients transferred into SUSTAIN-1, have been previously reported1,2,6,7 and were conducted in accordance with the Declaration of Helsinki, Good Clinical Practices, and applicable regulatory requirements. Protocols were approved by relevant institutional review boards and independent ethics committees. All patients provided written informed consent.1,2,6,7
SUSTAIN-1 was a double-blind, placebo-controlled, relapse-prevention trial that compared the efficacy of ESK versus placebo (PBO) nasal spray, both in conjunction with an OAD, in delaying relapse of depressive symptoms in patients with TRD who were in stable remission or stable response after 4 months of treatment with ESK plus OAD.1 SUSTAIN-2 was an open-label multicenter study that assessed the long-term safety and efficacy of ESK plus OAD in patients with TRD.7
The present analysis was limited to the 2 doses of ESK (56 and 84 mg) approved for use by the FDA in patients with TRD. Patients received ESK (56 or 84 mg) or PBO nasal spray (SUSTAIN-1 only), in conjunction with a newly initiated OAD. In SUSTAIN-1, after week 16 as part of the randomized withdrawal design, a subset of patients previously assigned to ESK were switched to PBO nasal spray and thus excluded from the current analysis after this time. Nasal spray treatment occurred twice weekly during the induction phase (weeks 1–4), once weekly during weeks 5–8, and thereafter once weekly or every other week in the optimization/maintenance phase, with dosing frequency individualized according to the severity of depressive symptoms (MADRS total score ≤ vs > 12) and tolerability.1,7 It was recommended that, on dosing days, patients take their OADs ≥ 3 hours after the ESK or PBO treatment session.
Safety Assessments
Patients were monitored for ≥ 90 minutes after ESK administration; afterward, they could leave the site if deemed clinically stable by the clinician. AEs were monitored and reported; safety assessments were conducted throughout the study. Clinician-reported AEs were categorized as mild, moderate, or severe per clinical judgment (Supplementary Table 1).
Vital signs, the Clinician-Administered Dissociative States Scale (CADSS), and the Modified Observer’s Assessment of Alertness/Sedation (MOAA/S) were assessed at baseline and at all treatment sessions (predose, 40 minutes, 1 hour [vital signs only], and 1.5 hours after dose). Abnormal CADSS and MOAA/S scores could be reported as AEs based on clinician judgment. Blood pressure (BP) readings were conducted at each treatment session, with elevations reported as AEs if clinically significant based on clinician judgment. The CADSS consists of 23 questions, each coded on a 5-point scale (0, not at all; 4, extreme), yielding a total score of 0–92.14,15 A CADSS total score of > 4 was used to indicate the presence of dissociative symptoms.15 Although the extent to which this score represents a clear marker for dissociation is debated,16 this cutoff has precedence in the literature and was determined as acceptable for use in the package labeling by FDA.8 Scores on the MOAA/S range from 0 (no response to painful stimulus) to 5 (readily responds to name spoken in normal tone).17 Any decrease in MOAA/S from predose (score < 5) indicated some degree of sedation. Additionally, acute hypertension was defined per protocol as systolic BP of ≥ 180 mm Hg and ≥ 20 mm Hg higher than baseline and/or diastolic BP of ≥ 105 mm Hg and ≥ 15 mm Hg higher than baseline. This analysis included any treatment sessions in which patients received ESK. We first looked for any discernable patterns in the occurrence of AEs over time following repeated administrations of ESK, stratified according to whether the respective AEs occurred once, twice, or not at all during the index week. Weeks 1 and 4 were set a priori as index weeks because they represent the beginning and end of the induction period, and previous reports have documented that the frequency and/or severity of at least some of the AEs decline over the first 4 weeks of treatment.7 We also examined whether the maximum reported AE severities during the index week provided an indication of the severities of AE recurrences.
Recurrence and severity of AEs were assessed from week 2–4, month 2 (weeks 5–8), months 3–6, and months 6–12. Study investigators rated AE severity as 0 (no AE), 1 (mild), 2 (moderate), or 3 (severe) based on the clinician’s judgment. To examine AEs in individual patients over time, only patients who received ≥ 1 ESK dose in the subsequent period were included in the analyses of each respective period.
The 5 most commonly reported treatment-emergent AEs occurring with ESK plus OAD (dissociation, dizziness, nausea, sedation, and vertigo) were evaluated, each having an incidence of ≥ 5% and at least a 2-fold greater incidence compared with PBO nasal spray plus an OAD.8 Observed increases in BP were also examined at each dosing session.
Patients who received medication to treat an emergent AE or prevent re-emergence of an AE were identified; however, given the low numbers, additional analysis to assess potential impact was not possible. Patients identified as having adequately controlled hypertension before initiation of treatment were instructed to take their hypertension medication before nasal spray dosing. Incidence of BP-related AEs was examined in patients with a history of hypertension according to whether they were taking medication to manage their hypertension.
RESULTS
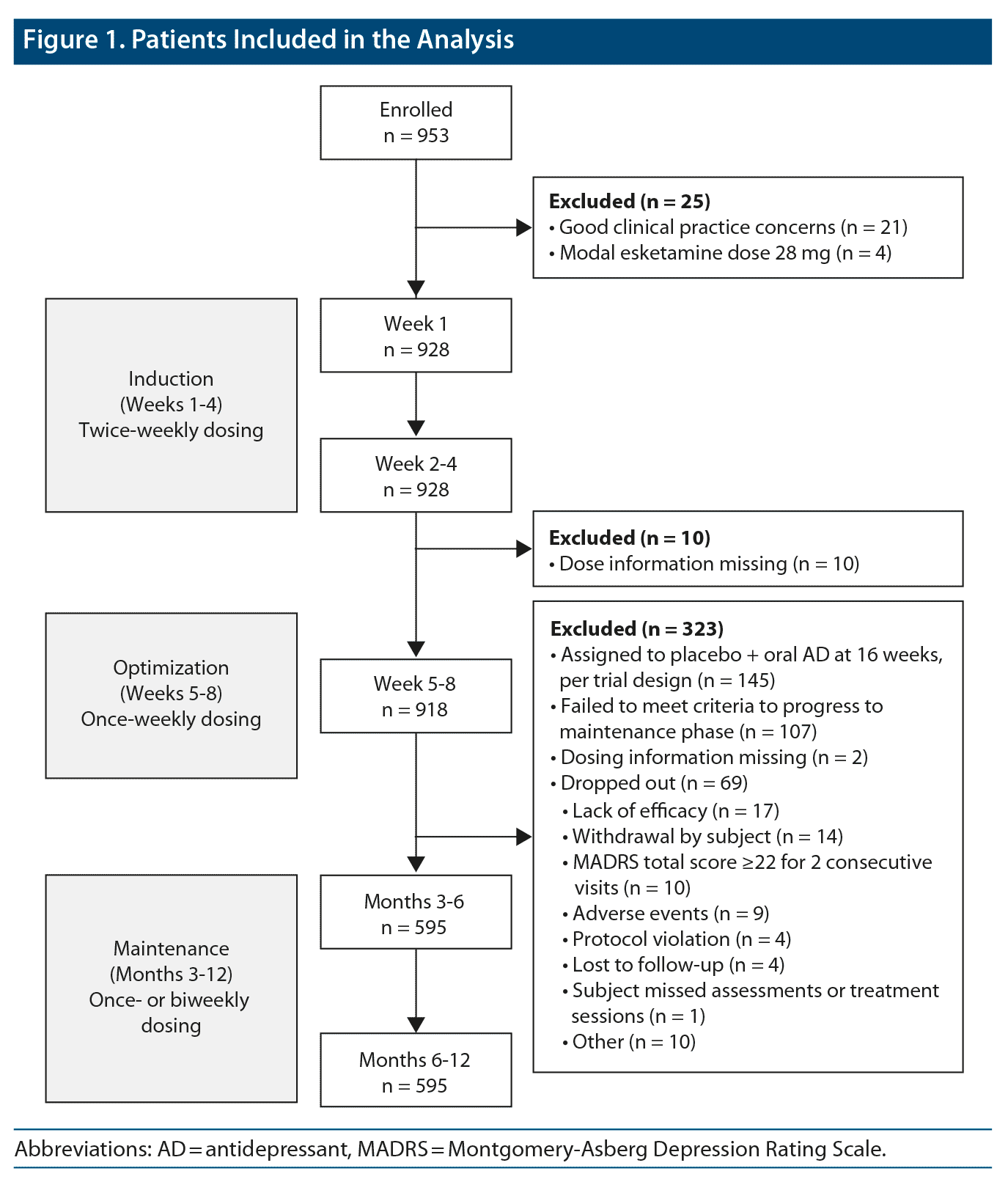
This analysis included 928 patients in weeks 1–4, 918 in weeks 5–8, and 595 in months 3–6 and 6–12 (Figure 1). The mean age of the patients was 46 years; approximately two-thirds were female (Supplementary Table 2). Discontinuations, including those due to AEs, were low during the first 4 weeks of treatment (5%–7%).1,7
AE Rates Observed by Frequency of AEs During Week 1
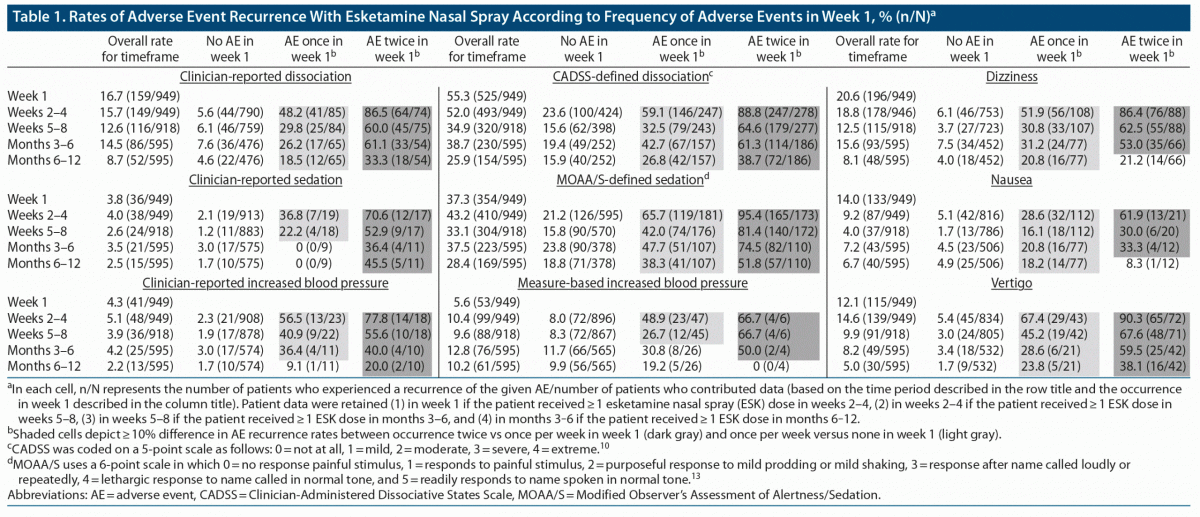
The more frequently an AE occurred during the first week of treatment, the higher was the percentage of patients who experienced recurrence in subsequent time periods (Table 1). For example, the rate of dissociation based on spontaneous AE reporting during weeks 2‒4 for the overall patient population was 15.7%. However, the corresponding rate for those in whom dissociation was reported twice as an AE during week 1 was 86.5% (64/74), whereas the rates in those for whom dissociation was reported once or not at all during week 1 were 48.2% (41/85) and 5.6% (44/790), respectively (Table 1). The same pattern (recurrence rates increasing with week 1 AE frequency) was observed for each examined AE and for corresponding rates of dissociation, sedation, and elevated BP based on standardized measures (Supplementary Figure 1).
AE Rates Observed by Frequency in Week 1 vs Frequency in Week 4
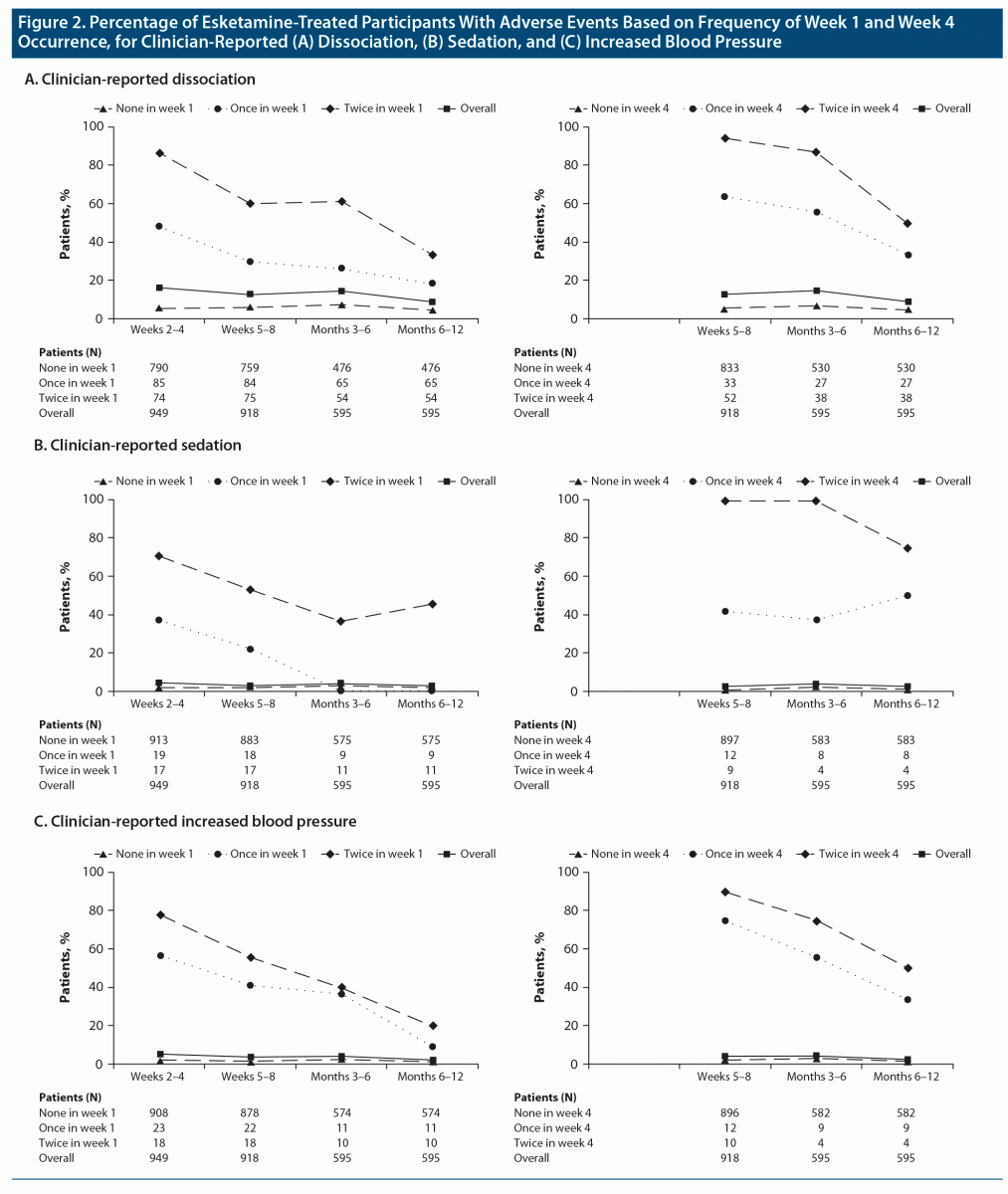
AE rates after week 4 appeared more like those observed in week 4 than those in week 1. When an AE did not occur in week 1 or week 4, there was little difference between the rates observed thereafter for clinician-reported AEs of dissociation, sedation, and increased BP (Figure 2); in these groups, there were few recurrences. Relationships for other AEs, including scale-based rates of dissociation, sedation, and acute hypertension, followed a similar pattern (Supplementary Figure 1), although the differences for acute hypertension were relatively small.
Does the Severity of an AE Experienced Early (ie, Week 1 or Week 4) Provide an Indication of AE Severity Later in Treatment?
The ability to explore if AE severity in weeks 1 or 4 could be used to inform subsequent AE severity was limited by the low number of patients with moderate or severe AEs. For all spontaneously reported AEs (except dissociation), < 5 patients for whom the respective AEs were reported as severe in weeks 1 or 4 had a recurrence reported as severe during any subsequent observation period. Likewise, for AEs other than dissociation, dizziness, vertigo, or nausea, < 10 patients for whom the AEs were reported in weeks 1 or 4 experienced recurrences of a moderately severe event.
AEs that recurred were generally mild or moderate in severity. Furthermore, the initial severity of an AE provided little indication of future severity, most likely due to minimal variance in AE severity. There were no consistent differences in the severity of respective AEs when comparing patients without a reported AE during weeks 1 or 4 versus those with mild AEs. When comparing patients whose AEs of dissociation were characterized as moderate (score: 1) or severe (score: 2) in week 1, for those reporting a recurrence in subsequent weeks, mean AE severity scores were 1.5 and 2.1 in weeks 2–4, 1.3 and 1.8 in weeks 5–8, 1.4 and 1.2 in months 3–6, and 1.5 and 1.6 in months 6–12, respectively. In comparison, the mean AE severity score reported in patients who experienced their first reported dissociation after week 1 was 1.3.
In patients with evidence of mild-to-moderate levels of dissociation per CADSS (total score ≤ 14),16 there were no obvious differences in the severity of subsequent spontaneously reported AEs of dissociation (mean scores: 1.2–1.4) across the different follow-up timeframes. Mean severity scores in patients with maximum CADSS total scores 15–24 and 24–42 were 1.3–2.1 across the different follow-up timeframes, and < 3 patients had a maximum CADSS score of > 42.
Finally, even when stratifying patients according to their most marked level of sedation observed during weeks 1 or 4, the average severity observed in recurrences was indicative of mild sedation (MOAA/S scores: 3.5–4.0), except for 1 patient with a mean recurrence score of 3.25, reflecting moderate sedation.
What Is the Impact of ESK Dose and Concomitant Medications on AEs?
Because flexible dosing (56 or 84 mg) was permitted in the first 2 weeks of the studies’ induction phases, patients were stratified by modal dose during the respective periods. A small dose effect was most pronounced with dissociation and dizziness (Supplementary Figure 2); however, the impact of dose on AE recurrence rates was of a much smaller magnitude than the effect of week 1 AE frequency. Among patients who exhibited the most frequently observed AEs during week 1, few received prophylactic or symptomatic treatment for these AEs in later treatment sessions (Supplementary Table 3). BP-related AEs were also examined in patients with a history of hypertension and stratified according to whether the hypertension was managed with medication. As with other AEs, the number of patients for whom BP-related AEs were reported was too small to draw meaningful conclusions (Supplementary Table 4).
DISCUSSION
This post hoc analysis of adults treated with ESK plus OAD for TRD revealed that patients who experienced one of the most commonly reported AEs once or twice during the first week of treatment were more likely to experience a recurrence of the same AE compared to those who did not. Overall, recurrence rates diminished over time. If an AE occurred in week 4, its recurrence thereafter was more closely linked to its week 4 frequency than its week 1 frequency. The restricted range of observed AE severity, with most being mild or moderate, limited the ability to predict the severity of recurring AEs.
These findings suggest that the likelihood of AE recurrence may increase along with the frequency with which the AE occurs early in treatment. Nevertheless, the severity for such recurrences is likely to be mild to moderate and tends to diminish over time. Machine-learning–based algorithms using more variables may outperform this practical single-indicator clinical approach,18 but these are typically not used in clinical practice due to complexity, lack of transparency of underlying criteria, and perceived additional effort.19 Although it is impossible to predict whether a particular adverse event will or will not occur in a patient, our results may help inform shared decision-making between patient and clinician during treatment with ESK, within the guidelines of the REMS.9 Because the original studies were not designed to formally evaluate potential factors impacting adverse event occurrence, it is unknown whether this occurrence is influenced by baseline patient factors, such as symptom severity, personality20 or psychiatric factors,21 or by the magnitude of improvement in depressive symptoms, but these relationships could be explored in future studies.
Differences between formally measured versus spontaneously reported rates of AEs of dissociation, sedation, and abnormally elevated BP are evident, with formally measured (scale-based) rates being higher than clinician-reported rates. These measurement approaches are inherently calibrated to different standards: clinicians were encouraged to report any AE they believed was clinically relevant or merited treatment, whereas the solicited formal measures assessed at each treatment session (eg, CADSS, MOAA/S, BP measurement) simply detect deviations from an accepted standard of “normal” values. Therefore, higher rates are more often revealed by the formal measures obtained by solicited reporting than by clinician report. Nevertheless, the same fundamental relationships between early and later tolerability were observed across measurement modalities.
A recent comprehensive analysis of cardiovascular safety from the phase 3 TRD program described BP elevations after administration of ESK plus OAD (in patients with no or controlled hypertension) as transient (maximum elevation within 40 minutes of ESK dosing; typically returning to predose range ≤ 1.5 hours after dosing), asymptomatic, self-limiting, not warranting rescue medications, and not associated with serious cardiovascular safety sequalae.13 Although lower in patients who had not previously experienced the same AE, rates of objectively measured dissociation, sedation, and elevated BP never reached zero; therefore, consistent with prescribing recommendations, patients who experienced one of these AEs should be monitored after dosing to ensure sedation and dissociation are resolved before discharge, and BP must be assessed before ESK administration, at approximately 40 minutes after receiving a dose, and subsequently as clinically warranted.8
Regarding study limitations, this was a post hoc analysis of pooled data from 2 long-term studies with differing designs (SUSTAIN-1, double-blind; SUSTAIN-2, open-label) where the minimum required monitoring period per protocol was 90 minutes, in contrast to the ≥ 2-hour post-administration monitoring required by the REMS. Study exclusion criteria may have skewed the sample to patients with fewer comorbidities than observed in real-world clinical settings. Analyses are limited to specific events as measured or reported by clinical trial sites during postdose monitoring. The consistency with which an AE is reported may vary over time and between sites. For instance, a clinician may consider an AE as having minimal clinical impact on a patient and thus be less likely to report it; conversely, an individual patient may find a given AE more troubling than other patients, even at mild levels, so a clinician’s threshold for reporting the AE may shift reactively over time. These dynamics would impact reported recurrence rates in opposite directions and are difficult to quantify. Few patients (0.8%–5%; Supplementary Table 3) received prophylactic or symptomatic treatment for AEs during the respective timeframes, suggesting that these treatments had minimal impact on the observed patterns.
Finally, because AE recurrence was central to this post hoc analysis, we prospectively chose to limit our sample to patients who had ≥ 1 treatment session in the “next” timeframe to ensure we were examining patients in whom AEs had an opportunity to recur. This raises the potential for completer selection bias, whereby a systematic difference in patients who do not continue through the various stages of treatment may be missed. Given the low discontinuation rate during the first month of treatment (5%–7% by week 4), it is probable this potential bias had minimal impact on the rates of AEs observed during the first 2 months of treatment. The design of SUSTAIN-1 (whereby patients who did not meet criteria for stable response or stable remission at week 16 did not continue, and half of those remaining who were originally assigned to ESK were transitioned to placebo nasal spray in the subsequent randomized withdrawal phase) prevented a substantial number of patients from being followed after month 4 of treatment.
Overall, this post hoc analysis provides additional information for prescribers that can assist in clinician-patient discussions regarding the tolerability of ESK, setting expectations regarding AEs, and the likelihood and potential management of their recurrence. Clinicians should initiate discussions with patients about possible AEs that may occur with ESK treatment while recognizing that AEs are rarely severe and become less frequent with ongoing treatment.
Submitted: November 9, 2021; accepted June 9, 2022.
Published online: September 19, 2022.
Relevant financial relationships: Drs Gogate, Kern Sliwa, and Starr are employees of Janssen Scientific Affairs, LLC. Dr Preskorn has, within the last year, received research grants (with funding payments made to the University of Kansas Medical Center Research Institute) from Acadia, Allergan, and Janssen. He has received compensation as a consultant for Alkermes, BioXcel, Janssen, Merck, Novartis, and Perception Neuroscience and as a speaker for Janssen and Sunovion. Dr Winokur received research grants (with funding payments made to Hartford Healthcare) from Otsuka, Allergan, Boehringer Ingelheim, Janssen, and NeuroRx. Drs Williamson, Daly, and Manera were employees of Janssen Scientific Affairs, LLC at the time the study was conducted.
Funding/support: This research was funded by Janssen Scientific Affairs, LLC.
Role of the sponsor: Employees of the sponsor, as noted in author contributions, were involved in study design; patient recruitment; data collection, analysis, or interpretation; and/or other aspects pertinent to the study. Authors had full access to the study data, were involved in writing and/or revising the manuscript, and had final responsibility for the decision to submit for publication. Medical writing assistance for this manuscript was also supported by the sponsor.
Previous presentation: Poster presented at the American Society of Clinical Pharmacology (ASCP) Annual Meeting; May 29–31, 2019; Scottsdale, AZ.
Acknowledgments: The authors thank the SUSTAIN 1 and 2 study investigators who originally conducted the study and enrolled the patients. The authors also thank Sarah Feeny, BMedSci, and Lynn Brown, PhD, of ApotheCom (UK and US) for medical writing and editorial services, which were funded by Janssen Scientific Affairs. Ms Feeny and Dr Brown have no conflicts of interest to declare.
Supplementary material: Available at Psychiatrist.com.
Clinical Points
- The 5 most common adverse events (AEs; dissociation, dizziness, nausea, sedation, vertigo) associated with esketamine nasal spray plus an oral antidepressant in patients with treatment-resistant major depressive disorder are more likely to recur after week 1 if they occurred more frequently during the first week of treatment.
- Most patients did not report AEs in week 1 and were stratified into the groups with the lowest recurrence risk.
- Recurrence of AEs after week 4 (the end of the induction period) was more closely related to the frequency of the same AEs during week 4 than during week 1.
- The reported severity of dissociation, dizziness, nausea, sedation, vertigo, and increased blood pressure was mostly mild and rarely severe, in terms of both initial incidence and recurrence.
References (21)

- Daly EJ, Trivedi MH, Janik A, et al. Efficacy of esketamine nasal spray plus oral antidepressant treatment for relapse prevention in patients with treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2019;76(9):893–903. PubMed CrossRef
- Fedgchin M, Trivedi M, Daly EJ, et al. Efficacy and safety of fixed-dose esketamine nasal spray combined with a new oral antidepressant in treatment-resistant depression: results of a randomized, double-blind, active-controlled study (TRANSFORM-1). Int J Neuropsychopharmacol. 2019;22(10):616–630. PubMed CrossRef
- Fu DJ, Ionescu DF, Li X, et al. Esketamine nasal spray for rapid reduction of major depressive disorder symptoms in patients who have active suicidal ideation with intent: double-blind, randomized study (ASPIRE I). J Clin Psychiatry. 2020;81(3):19m13191. PubMed CrossRef
- Ionescu DF, Fu DJ, Qiu X, et al. Esketamine nasal spray for rapid reduction of depressive symptoms in patients with major depressive disorder who have active suicide ideation with intent: results of a phase 3, double-blind, randomized study (ASPIRE II). Int J Neuropsychopharmacol. 2021;24(1):22–31. PubMed
- Ochs-Ross R, Daly EJ, Zhang Y, et al. Efficacy and safety of esketamine nasal spray plus an oral antidepressant in elderly patients with treatment-resistant depression-TRANSFORM-3. Am J Geriatr Psychiatry. 2020;28(2):121–141. PubMed CrossRef
- Popova V, Daly EJ, Trivedi M, et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am J Psychiatry. 2019;176(6):428–438. PubMed CrossRef
- Wajs E, Aluisio L, Holder R, et al. Esketamine nasal spray plus oral antidepressant in patients with treatment-resistant depression: assessment of long-term safety in a phase 3, open-label study (SUSTAIN-2). J Clin Psychiatry. 2020;81(3):19m12891. PubMed CrossRef
- Spravato (esketamine) nasal spray, CIII. Package insert. Janssen Pharmaceuticals, Inc.; July 2020:15.
- Risk Evaluation and Mitigation Strategy (REMS) Document. Spravato (esketamine hydrochloride) REMS Program. FDA website. https://www.accessdata.fda.gov/drugsatfda_docs/rems/Spravato_2019_06_25_REMS_Document.pdf. Updated June 2019. Accessed February 9, 2022.
- Pereira S, Brennan E, Patel A, et al. Managing dissociative symptoms following the use of esketamine nasal spray: a case report. Int Clin Psychopharmacol. 2021;36(1):54–57. PubMed CrossRef
- Starr L, Winokur A, Daly E, et al. Adverse events affecting readiness for discharge from the clinical setting after esketamine nasal spray administration. Presented at the 32nd annual US Psych Congress; October 3–6, 2019; San Diego, CA.
- Janssen Research & Development LLC. FDA Advisory Committee Briefing Document. Esketamine nasal spray for patients with treatment resistant depression. JNJ-54135419 (esketamine). https://www.fda.gov/media/121377/download. Updated 2019. Accessed March 8, 2022.
- Doherty T, Wajs E, Melkote R, et al. Cardiac safety of esketamine nasal spray in treatment-resistant depression: results from the clinical development program. CNS Drugs. 2020;34(3):299–310. PubMed CrossRef
- Bremner JD, Krystal JH, Putnam FW, et al. Measurement of dissociative states with the Clinician-Administered Dissociative States Scale (CADSS). J Trauma Stress. 1998;11(1):125–136. PubMed CrossRef
- Bremner JD. The Clinician Administered Dissociative States Scale (CADSS): instructions for administration. https://docplayer.net/41963296-Instructions-for-administration.html. 2021. Accessed March 8, 2022.
- Williamson D, Turkoz I, Wajs E, et al. Linking adverse events of dissociation reported with esketamine dosing by clinician-perceived severity and CADSS descriptors. Presented at the 15th Annual Scientific Meeting of the International Society for CNS Clinical Trials and Methodology (ISCTM); February 19–21, 2019; Washington, DC.
- Chernik DA, Gillings D, Laine H, et al. Validity and reliability of the Observer’s Assessment of Alertness/Sedation Scale: study with intravenous midazolam. J Clin Psychopharmacol. 1990;10(4):244–251. PubMed
- Kautzky A, Dold M, Bartova L, et al. Refining prediction in treatment-resistant depression: results of machine learning analyses in the TRD III sample. J Clin Psychiatry. 2018;79(1):16m11385. PubMed CrossRef
- Benda NC, Das LT, Abramson EL, et al. “How did you get to this number?” stakeholder needs for implementing predictive analytics: a pre-implementation qualitative study. J Am Med Inform Assoc. 2020;27(5):709–716. PubMed CrossRef
- Kern A, Kramm C, Witt CM, et al. The influence of personality traits on the placebo/nocebo response: a systematic review. J Psychosom Res. 2020;128:109866. PubMed CrossRef
- Pennybaker SJ, Luckenbaugh DA, Park LT, et al. Ketamine and psychosis history: antidepressant efficacy and psychotomimetic effects postinfusion. Biol Psychiatry. 2017;82(5):e35–e36. PubMed CrossRef
This PDF is free for all visitors!