Objective: While doctors often increase the dose of an antipsychotic when there is insufficient response, there is limited evidence that this intervention is any better than waiting longer on the lower dose. We put the proposition to test.
Method: In this 4-week, double-blind, randomized controlled trial conducted in psychiatric care from September 2012 to March 2015, 103 patients with schizophrenia (ICD-10) who did not respond to olanzapine 10 mg/d or risperidone 3 mg/d were randomly allocated to a dose-increment or -continuation group. In the increment group, antipsychotic doses were doubled for 4 weeks, whereas in the continuation group, doses were not changed. Completion rate (primary outcome measure); changes in psychopathology, function, and extrapyramidal symptoms; and response rate were compared between the groups. The relationship between baseline plasma antipsychotic concentrations and changes in psychopathology was examined.
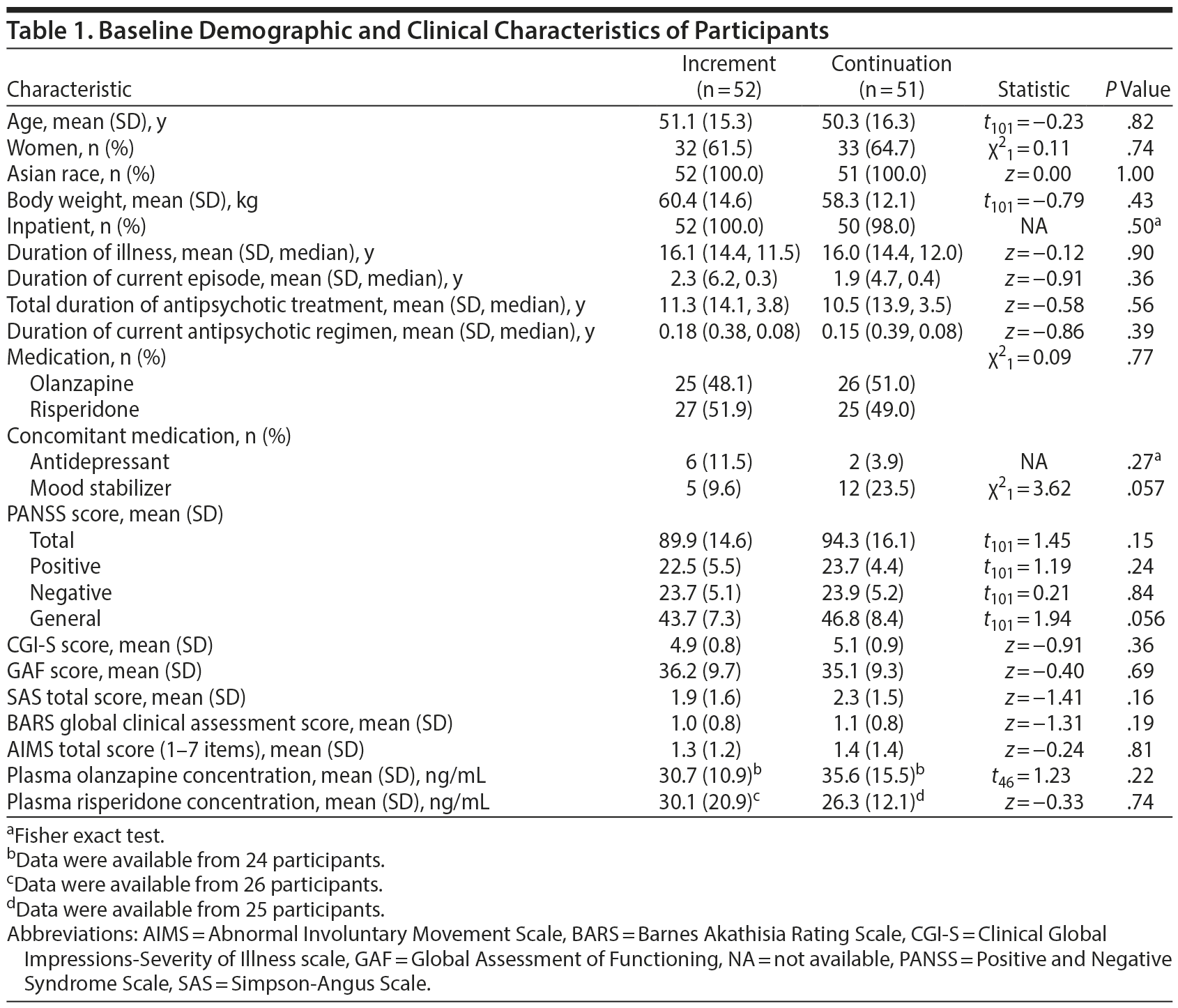
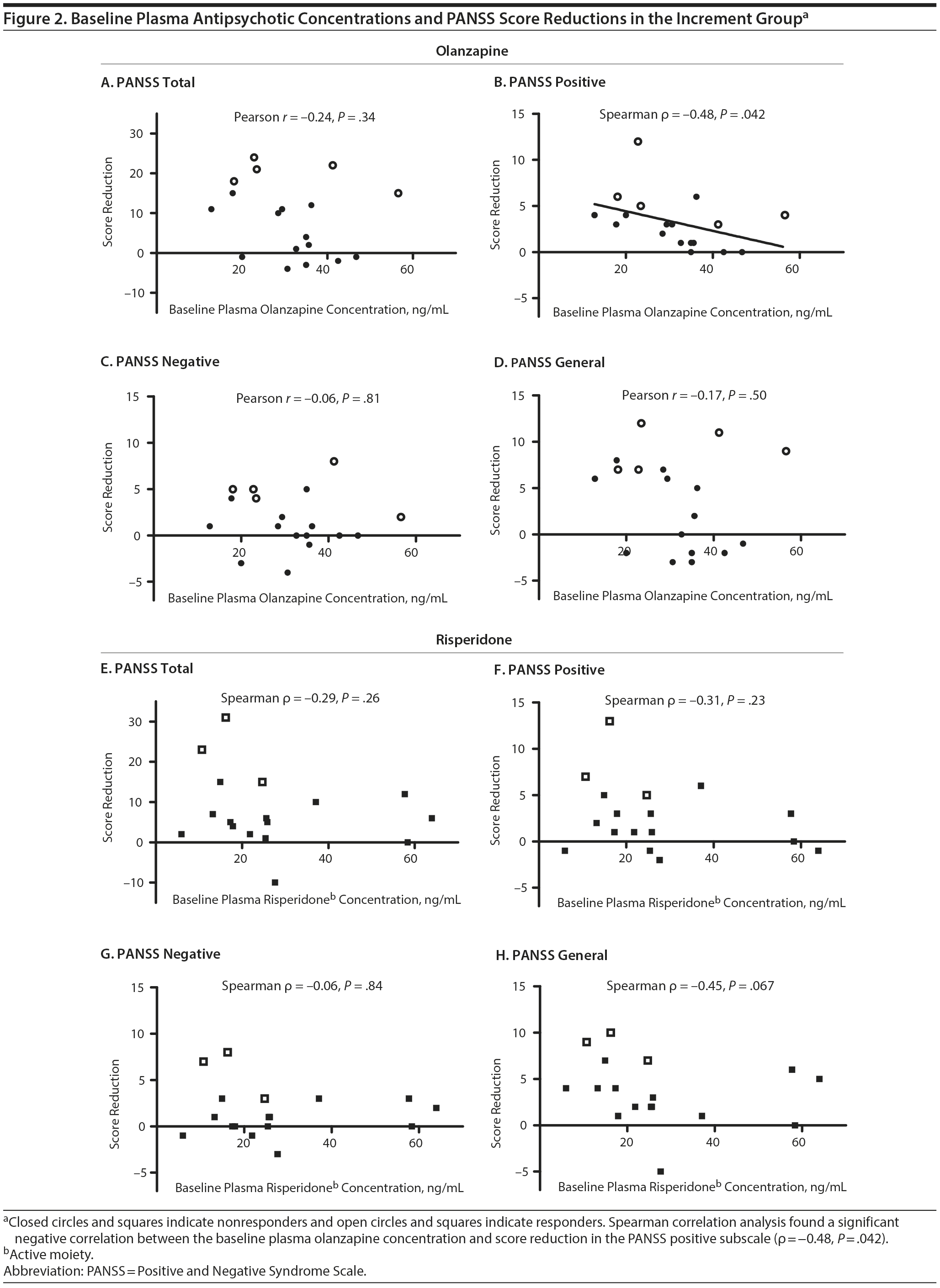
Results: The completion rate was significantly lower in the increment group than in the continuation group (69.2% [36/52] vs 86.3% [44/51], P = .038). No significant superiority was observed in any of the outcome measures in the increment group compared to the continuation group, except the Positive and Negative Syndrome Scale (PANSS) positive subscale score change in intention-to-treat analysis. Those with lower plasma concentrations of olanzapine on their initial treatment showed a greater improvement on the PANSS positive subscale when their dose was increased (P = .042).
Conclusions: As a general strategy, patients with schizophrenia failing to respond to moderate antipsychotic doses may not benefit from an increase in dose. The possibility of benefit in those whose plasma antipsychotic concentrations at baseline are still low cannot be ruled out.
Trial Registration: UMIN.ac.jp/ctr/index.htm identifier: UMIN000008667
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
CME Background
Articles are selected for credit designation based on an assessment of the educational needs of CME participants, with the purpose of providing readers with a curriculum of CME articles on a variety of topics throughout each volume. Activities are planned using a process that links identified needs with desired results.
To obtain credit, read the article, correctly answer the questions in the Posttest, and complete the Evaluation. A $5 processing fee will apply.
CME Objective
After studying this article, you should be able to:
• Check plasma antipsychotic concentrations before increasing dose in patients with schizophrenia who have had insufficient treatment response
Accreditation Statement
The CME Institute of Physicians Postgraduate Press, Inc., is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Credit Designation
The CME Institute of Physicians Postgraduate Press, Inc., designates this journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Note: The American Academy of Physician Assistants (AAPA) accepts certificates of participation for educational activities certified for AMA PRA Category 1 Credit™ from organizations accredited by ACCME or a recognized state medical society. Physician assistants may receive a maximum of 1 hour of Category I credit for completing this program.
Date of Original Release/Review
This educational activity is eligible for AMA PRA Category 1 Credit™ through October 31, 2018. The latest review of this material was September 2016.
Financial Disclosure
All individuals in a position to influence the content of this activity were asked to complete a statement regarding all relevant personal financial relationships between themselves or their spouse/partner and any commercial interest. The CME Institute has resolved any conflicts of interest that were identified. In the past year, Alan J. Gelenberg, MD, has been a consultant for Zynx Health, has been a stock shareholder of Healthcare Technology Systems, and has participated in antitrust litigation on behalf of GlaxoSmithKline. No member of the CME Institute staff reported any relevant personal financial relationships. Faculty financial disclosure appears at the end of the article.
Increasing Versus Maintaining the Dose of Olanzapine or Risperidone
in Schizophrenia Patients Who Did Not Respond to a Modest Dosage:
A Double-Blind Randomized
Controlled Trial
ABSTRACT
Objective: While doctors often increase the dose of an antipsychotic when there is insufficient response, there is limited evidence that this intervention
is any better than waiting longer on the lower dose. We put the proposition
to test.
Method: In this 4-week, double-blind, randomized controlled trial conducted in psychiatric care from September 2012 to March 2015, 103 patients with schizophrenia (ICD-10) who did not respond to olanzapine 10 mg/d or risperidone 3 mg/d were randomly allocated to a dose-increment or -continuation group. In the increment group, antipsychotic doses were doubled for 4 weeks, whereas in the continuation group, doses were not changed. Completion rate (primary outcome measure); changes in psychopathology, function, and extrapyramidal symptoms; and response rate were compared between the groups. The relationship between baseline plasma antipsychotic concentrations and changes in psychopathology was examined.
Results: The completion rate was significantly lower in the increment group than in the continuation group (69.2% [36/52] vs 86.3% [44/51], P = .038). No significant superiority was observed in any of the outcome measures in the increment group compared to the continuation group, except the Positive and Negative Syndrome Scale (PANSS) positive subscale score change in intention-to-treat analysis. Those with lower plasma concentrations of olanzapine on their initial treatment showed a greater improvement on the PANSS positive subscale when their dose was increased (P = .042).
Conclusions: As a general strategy, patients with schizophrenia failing to respond to moderate antipsychotic doses may not benefit from an increase in dose. The possibility of benefit in those whose plasma antipsychotic concentrations at baseline are still low cannot be ruled out.
Trial Registration: UMIN.ac.jp/ctr/index.htm identifier: UMIN000008667
J Clin Psychiatry 2016;77(10):1381–1390
dx.doi.org/10.4088/JCP.15m10490
© Copyright 2016 Physicians Postgraduate Press, Inc.
aDepartment of Neuropsychiatry, Keio University School of Medicine, Tokyo, Japan
bDepartment of Psychiatry, Inokashira Hospital, Tokyo, Japan
cDivision of Clinical Pharmacology, Indiana University School of Medicine, Indianapolis
dGeriatric Mental Health Program, Centre for Addiction and Mental Health, Toronto, Ontario, Canada
eDepartment of Psychiatry, University of Toronto, Toronto, Ontario, Canada
fDepartment of Psychosis Studies, Institute of Psychiatry, King’s College, London, United Kingdom
*Corresponding author: Hiroyuki Uchida, MD, PhD, Department of Neuropsychiatry, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo, 160-8582, Japan ([email protected]).
Antipsychotic drugs play an indispensable role in the treatment of schizophrenia.1 Based on clinical trials and brain-imaging data, recommended doses for these drugs are now available but often span a 100% range or more (eg, 10–20 mg/d for olanzapine and 2–6 mg for risperidone).2 These ranges are derived from some fixed-dose trials,3–5 and while all doses within the range usually show efficacy, there is little evidence to suggest that moving from a low dose within the recommended range to a higher dose brings any further clinical benefit. Yet, the question is of critical importance, as increasing dose is the most commonly undertaken clinical maneuver upon suboptimal response to a modest dosage.
Few studies6–10 thus far have systematically addressed this question. In general, these trials suggest that dose increment does not yield any additional clinical gain compared to continuation of the standard doses.11 However, the data are limited to fluphenazine, haloperidol, quetiapine, and ziprasidone, and more frequently prescribed antipsychotics such as olanzapine and risperidone have not yet been tested. Moreover, the dose ranges used in those studies were too high and do not represent current clinical practice (ie, fluphenazine 80 mg/d, haloperidol 12 mg/d, quetiapine 1,144 and 1,200 mg/d, and ziprasidone 320 mg/d). In addition, except for one ziprasidone study,10 clinical outcomes have not been explored in terms of blood antipsychotic concentrations, which more precisely reflect the exposure to antipsychotic drugs than oral dosage given wide interindividual differences in pharmacokinetic parameters.12 Thus, such a paucity of data still limits our dosing strategies for the treatment of schizophrenia.13
We therefore conducted a double-blind, randomized controlled trial to compare the efficacies between increasing antipsychotic dose and staying on the same dose in patients with schizophrenia who did not respond to moderate doses of olanzapine or risperidone. Furthermore, we evaluated the impact of baseline plasma antipsychotic concentrations on treatment outcomes.
METHOD
Study Design and Setting
This 4-week, double-blind, randomized controlled trial was conducted at Inokashira Hospital in Japan from September 2012 to March 2015. This trial was approved by the institutional review board of the hospital and registered at UMIN Clinical Trials Registry in August 2012 (UMIN.ac.jp/ctr/index.htm identifier: UMIN000008667). Written informed consent was obtained from all of the participants after a full explanation of the study was provided.
Participants
Inclusion criteria of participants were as follows: (1) inpatients or outpatients who met the criteria for schizophrenia, schizoaffective disorder, or persistent delusional disorder according to the International Classification of Diseases, Tenth Revision (ICD-10),14 which is widely used in Japan; (2) patients who had been receiving olanzapine 10 mg/d or risperidone 3 mg/d for ≥ 4 weeks in clinical settings; (3) a total score of ≥ 60 on the Positive and Negative Syndrome Scale (PANSS),15 ≥ 3 on the Clinical Global Impressions-Severity of Illness scale (CGI-S),16 and ≤ 70 on the Global Assessment of Functioning (GAF),17 (4) age of 20 years or older, and (5) ability to provide informed consent. Exclusion criteria were (1) concomitant use of another antipsychotic drug within the past 4 weeks, (2) history of nonresponse or intolerability to olanzapine 20 mg/d or risperidone 6 mg/d, (3) presence of active suicidal ideations or past suicide attempts, and (4) presence of severe physical diseases.
Procedures
Enrolled participants were randomly allocated to 1 of the 2 treatment groups in a 1:1 ratio by simple randomization stratified by their antipsychotic type (ie, olanzapine or risperidone) and treatment setting (ie, inpatient or outpatient). The person who was independent of this study in the central office prepared a piece of paper on which 1 of the assigned group was designated according to a computer-generated randomization list, inserted it into an envelope on which a participant ID number was written, and sealed it. Upon registration of each participant, 1 of the investigators opened the envelope that corresponded to the participant’s ID, and the person who prepared the envelopes confirmed that the envelopes were appropriately opened. In the dose-increment group, the ongoing antipsychotic dose was increased to 20 mg/d for olanzapine or 6 mg/d for risperidone followed by a 4-week observation, whereas in the dose-continuation group, the ongoing antipsychotic drug dose (ie, olanzapine 10 mg/d or risperidone 3 mg/d) was kept the same for 4 weeks. These doses were selected with a reference to the upper limit of the suggested dose ranges in the Texas Medication Algorithm Project (ie, 10–20 mg/d for olanzapine and 2–6 mg/d for risperidone).2 During the 4-week observation, all antipsychotic drugs were provided in identical powder form in amount and color with lactose added (ie, white powder of 0.6 g for participants receiving risperidone or its placebo and yellow powder of 2.5 g for those receiving olanzapine or its placebo). Thus, the participants were blinded to their allocated intervention. Other psychotropic drugs prescribed at baseline were kept constant or only reduced as clinically appropriate. When clinically indicated, lorazepam, zolpidem (for anxiety and insomnia), and biperiden (for extrapyramidal symptoms) were allowed.
Assessment Measures
The following assessments were performed by assessors who were blinded to the allocation: PANSS, CGI-S, GAF, Simpson-Angus Scale (SAS),18 Barnes Akathisia Rating Scale (BARS),19 and Abnormal Involuntary Movement Scale (AIMS)18 at baseline and week 4, and Clinical Global Impressions-Improvement scale (CGI-I)16 at week 4. Plasma samples were collected for the measurement of concentrations of olanzapine or risperidone active moiety (risperidone plus 9-hydroxyrisperidone) more than 12 hours’ postdose at baseline and week 4. Plasma concentrations of olanzapine and risperidone and 9-hydroxyrisperidone were assayed in heparinized plasma using LC/MS/MS (liquid chromatography with tandem mass spectrometry detection) at the Centre for Addiction and Mental Health in Toronto, with a limit of quantitation of 2.3 ng/mL, 0.82 ng/mL, and 2.13 ng/mL, respectively. The following information was also collected: concomitant medications, intervals between the last dose and blood draw, and demographic and clinical information including age, diagnosis, sex, race, weight, and duration of illness. Medication adherence was confirmed by plasma antipsychotic concentrations.
Population Pharmacokinetic Analysis
In case baseline plasma samples were taken before 12 hours postdose, plasma antipsychotic concentrations at trough were estimated for each individual with 2 samples taken at different points in time (ie, the sample taken before 12 hours postdose at baseline and the sample taken at week 4) using the established population pharmacokinetic models.20,21 The precision and reliability of this estimation has recently been confirmed in our population pharmacokinetic studies.22,23 The nonlinear mixed-effect models for olanzapine and risperidone active moiety were previously established using the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) data.24,25 These original studies used to establish the population pharmacokinetic models comprised 1,527 olanzapine concentrations from 523 participants and 1,236 risperidone and 9-hydroxyrisperidone concentrations from 490 participants. Both compounds were adequately described using a 1-compartment linear model with first-order absorption. The previously established models utilized exponentiated or log-normal interindividual variability on each pharmacokinetic parameter; a mixture distribution to assign the trimodal distribution of clearance, as cytochrome P450 2D6 genotype was not available for risperidone; an age effect on clearance of the 9-hydroxyrisperidone moiety; and sex, race, and age effects on olanzapine disposition.
Statistical Analysis
Baseline demographic and clinical characteristics were compared between the increment and continuation groups by the Pearson χ2 test or the Fisher exact test for categorical variables and by the Student t test or the Mann-Whitney U test for continuous variables, as appropriate. The primary outcome measure was the rate of participants who completed the 4-week treatment, since our main interest was to confirm the feasibility of antipsychotic dose increment. The secondary outcome measures were PANSS total score changes from baseline to week 4 and proportions of participants who achieved response, defined as a ≥ 25% reduction in the PANSS total score from baseline. Values of interests in treatment outcomes were compared between the 2 groups by the Pearson χ2 test or the Fisher exact test for categorical variables and by the Student t test or the Mann-Whitney U test for continuous variables on intention-to-treat (ITT) or per-protocol basis. Baseline values were also used as end point values for dropouts in ITT analysis, whereas only completers were analyzed in per-protocol analysis. Logistic regression analysis was performed to evaluate associations between the successful study completion and the following variables: dose allocation, age (ie, < 51 years or ≥ 51 years), gender, duration of total antipsychotic treatment (ie, < 0.5 years or ≥ 0.5 years), baseline PANSS total score (ie, < 95 or ≥ 95), and antipsychotic drugs used. In completers in each group, Pearson or Spearman correlation coefficients were calculated to examine the relationship between the baseline plasma antipsychotic concentrations and reductions in the PANSS total, positive, negative, and general subscale scores. Those analyses conducted for the whole sample were also repeated for a subgroup of relatively “fresh” participants who had been treated with antipsychotic drugs for less than 6 months. In addition, those analyses were also conducted for a subgroup of participants with their baseline plasma antipsychotic concentrations below the optimal range recommended in the AGNP (Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie) consensus guidelines26 (ie, < 20 ng/mL for both olanzapine and risperidone). The Shapiro-Wilk test was used to assess whether the data had a normal distribution. A 2-tailed P value of < .05 was considered statistically significant for all tests. All statistical analyses were conducted using the Statistical Package for the Social Sciences (SPSS), version 22.0, for Windows (IBM Corporation, Armonk, New York).
RESULTS
Patient Disposition and Characteristics
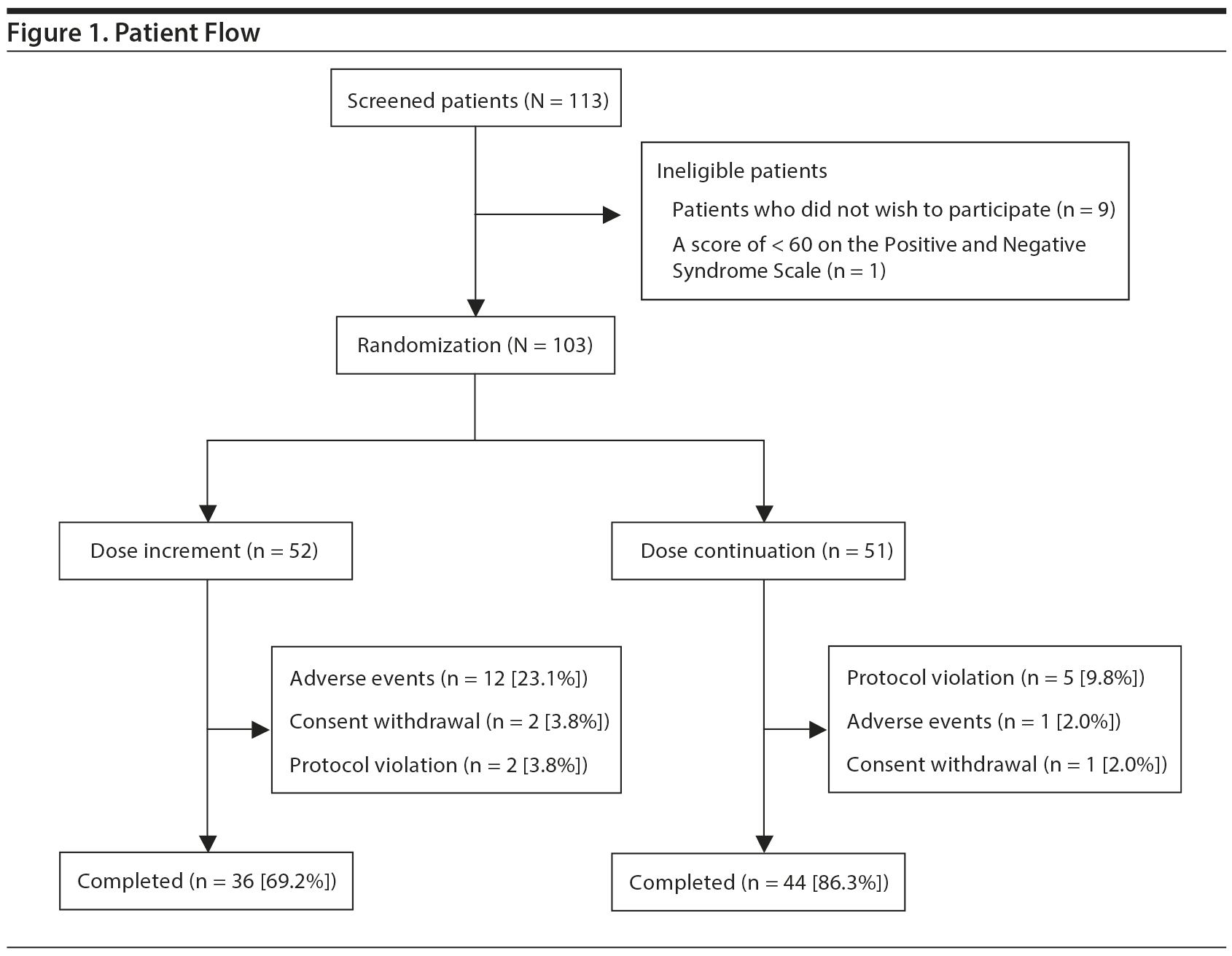
Of 113 eligible patients, 103 participants were enrolled and randomly assigned to either the increment group (n = 52) or the continuation group (n = 51) (Figure 1). Baseline demographic and clinical characteristics of the participants are shown in Table 1.
Outcome Measures
The completion rate was significantly higher in the continuation group than in the increment group (86.3% vs 69.2%) (Table 2). The logistic regression analysis found that only the same dose continuation was significantly associated with successful study completion (OR = 3.02; 95% CI, 1.08–8.47; P = .036; reference: dose increase) among the factors examined. Reasons for premature withdrawal are shown in Figure 1. Adverse events resulting in dropout were sedation (n = 8: olanzapine [n = 1] and risperidone [n = 7]), akathisia (n = 1, olanzapine), dysphagia (n = 1, risperidone), orthostatic hypotension (n = 1, olanzapine), and dyspnea (n = 1, olanzapine) in the increment group and akathisia (n = 1, olanzapine) in the continuation group. No serious adverse events were observed in both groups.
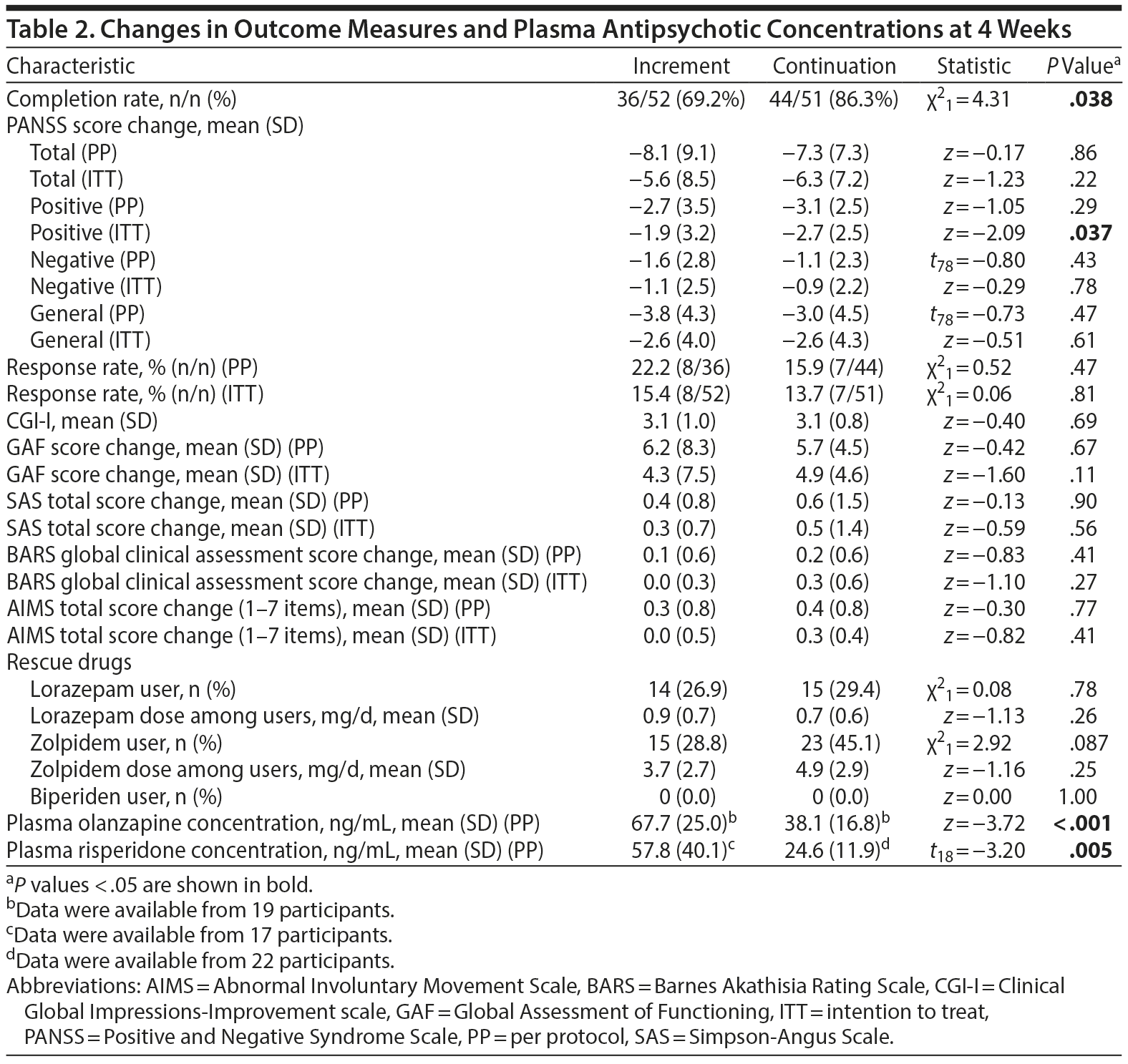
No statistically significant differences were found in any of the other outcome measures, including the response rate, between the 2 groups other than the PANSS positive subscale score change in ITT analysis (Table 2).
Plasma Concentrations and Outcome Measures
Thirty-five of 36 completers in the increment group provided blood samples both at baseline and at week 4, while 1 participant did not wish to provide the sample at baseline. Since 4 participants provided blood samples before 12 hours from the last dose, plasma drug concentrations at trough were individually estimated using the population pharmacokinetic models. Plasma antipsychotic concentrations at week 4 are shown in Table 2. In the increment group, there was a significant negative correlation between the baseline plasma antipsychotic concentration and score reduction in the PANSS positive subscale for olanzapine (Figure 2). In the continuation group, no statistically significant correlations were found between baseline plasma antipsychotic concentrations and reductions in the PANSS total or any subscale score.
Outcomes in a Subgroup of Participants
With < 6 Months of Antipsychotic Treatment
Baseline demographic and clinical characteristics of the 38 participants who had been treated with antipsychotic drugs for less than 6 months are shown in Supplementary eTable 1 (available at PSYCHIATRIST.COM). Whereas the completion rate was significantly higher in the continuation group than in the increment group (95.0% vs 61.1%), the response rate was significantly higher in the increment group than in the continuation group in per-protocol analysis (54.5% vs 15.8%) (Table 3). There were no statistically significant differences in any of the other outcome measures between those 2 groups.
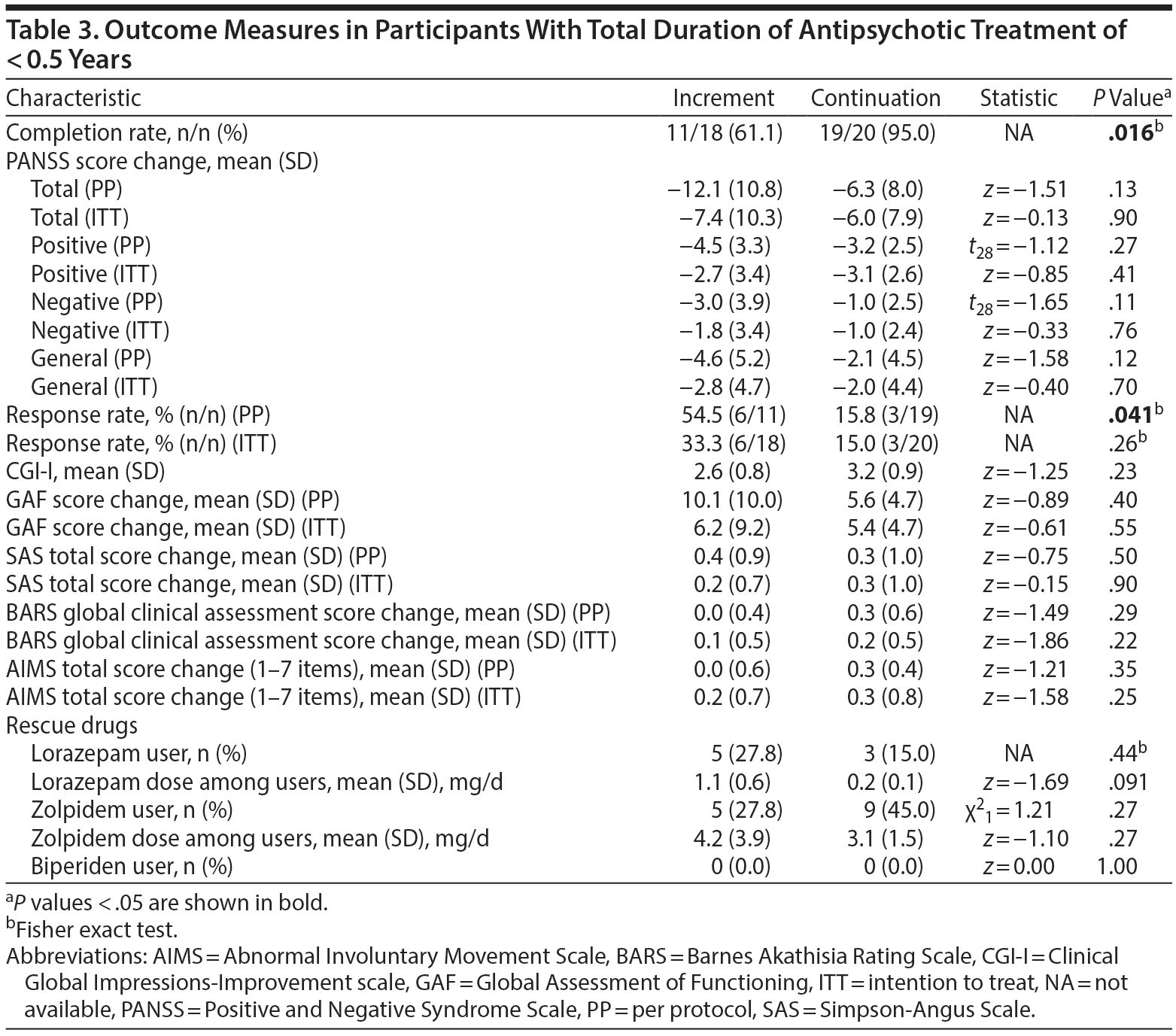
Outcomes in a Subgroup of Participants
With Suboptimal Plasma Antipsychotic
Concentrations at Baseline
Baseline demographic and clinical characteristics of the 29 participants with baseline plasma olanzapine or risperidone concentrations of < 20 ng/mL are shown in Supplementary eTable 2. There were no statistically significant differences in any of the outcome measures between the 2 groups, although the dose-increment group demonstrated numerically greater score reductions in the PANSS (Supplementary eTable 3).
DISCUSSION
At a group level, dose increment was not superior to dose continuation with respect to any of the psychopathological and functional measures, and the former was even inferior in terms of the completion rate in patients who failed to respond to moderate dosage of olanzapine and risperidone. Additionally, lower plasma olanzapine concentrations at baseline were associated with a greater improvement in the PANSS positive symptom subscale in the increment group. These findings, overall, might argue against any additional benefit of increasing the dose in cases of nonresponse to a moderate dosage of olanzapine and risperidone, but they do point to the potential of dose increment to improve positive symptoms for patients whose antipsychotic exposure is still below the optimal range.
The antipsychotic dose increment failed to yield any additional clinical gains; rather, it resulted in more frequent dropouts. This observation may suggest that another strategy, including switching or augmenting antipsychotic drugs, should be considered in case of insufficient response to a moderate dosage of antipsychotic drug. However, it should be interpreted with caution in light of the greater improvement in positive symptoms that we observed in those whose baseline antipsychotic exposure was low. The relationship between the oral dose of antipsychotic drugs and dopamine D2 receptor occupancy usually fits a 1-site binding model. Baseline doses of olanzapine 10 mg/d and risperidone 3 mg/d correspond to the relatively flat part of the nonlinear curve.27,28 Thus, dose increases would be expected to have a minimal impact on central occupancy and clinical response in general. In other words, the effectiveness of antipsychotic drugs may reach a plateau with those doses. On the other hand, a subgroup of patients whose antipsychotic exposure is still low due to their pharmacokinetic variations and who still fall on the steep part of the curve with the above-mentioned dosage may exhibit large changes in drug occupancy and therapeutic effects. These results are congruent with the finding from the ziprasidone study10 in which serum drug concentration changes following dose increase were correlated with improvements in the PANSS positive symptom subscale at a trend level but not with other symptom domains.
In the present study, lower plasma olanzapine concentrations at baseline were associated with greater improvement, following antipsychotic dose increase in positive symptoms, but not in negative symptoms or general psychopathology. While antipsychotic effects for positive symptoms have been shown to be robust, their impact on other symptom domains appears relatively limited.29,30 Moreover, antipsychotic treatment could result in secondary negative symptoms31 and neuroleptic dysphoria32 that are often difficult to be differentiated from the primary negative symptoms. Furthermore, high doses of antipsychotic drugs can even be detrimental to negative and cognitive symptoms.33,34 Our results may be interpreted as a ceiling effect of antipsychotic treatment for negative symptoms and general psychopathology.
In light of wide interindividual differences in responsiveness to antipsychotic drugs, we would like to refrain from simply stating, “Do not increase the dose in case of insufficient treatment response.” Rather, our results underscore the importance of individualized dosing strategy with therapeutic drug monitoring. It appears fair to suggest that dose increment may be considered for patients who present with positive symptoms and whose plasma antipsychotic concentrations are still low. Furthermore, potential benefits of such a dose-increase strategy may be especially true for patients who recently started antipsychotic treatment, although it could also result in more dropouts. While these preliminary notions have to be confirmed in large prospective studies, the conventional dosing strategy may need to be revisited, as oral dose alone cannot be regarded as a reliable measure of antipsychotic exposure.12
This study has several limitations. First, the sample size was small. Moreover, no sample size calculation was performed since the trial was conducted as a hypothesis-generating study. Second, the duration of 4 weeks may have been too short to compare the effectiveness and the safety of the 2 dosing strategies, although it has been shown that this duration is adequate to evaluate antipsychotic response.35 Third, since olanzapine and risperidone were the only drugs included, all participants were Japanese, 99.0% of the participants were inpatients, and the study sample included only patients who did not respond to moderate doses of olanzapine or risperidone, any extrapolation of these findings to other antipsychotic drugs or other populations must be made with caution. Fourth, heterogeneity of the sample should be taken into account. For example, the mean duration of current episode ranged from 0.03 years to 40 years, with a median of 0.4 years. Including both acute and chronic patients may have resulted in the overall low response rate as well as failure to find differences. Fifth, mixed-effect models for repeated measures were not used. They would have been ideal but were not employed in the present study since assessments were conducted only at the baseline and end point. Finally, corrections for multiple comparisons would have been ideal from a statistical perspective.
In conclusion, increasing the dose of an antipsychotic is a very common clinical intervention, especially when the dose is near the lower end of the standard range. We find no evidence to support this strategy in general. However, our findings do raise the possibility that the strategy may be a reasonable treatment option for those whose antipsychotic exposure is still insufficient. These findings emphasize the relevance of therapeutic drug monitoring to devise more effective dosing strategy on an individual basis for better management of schizophrenia.
Submitted: October 25, 2015; accepted February 11, 2016.
Drug names: biperiden (Akineton), fluphenazine (Prolixin, Permitil, and others), haloperidol (Haldol and others), lorazepam (Ativan and others), olanzapine (Zyprexa and others), quetiapine (Seroquel and others), risperidone (Risperdal and others), ziprasidone (Geodon and others), zolpidem (Ambien, Edluar, and others).
Disclosure of off-label usage: The authors have determined that, to the best of their knowledge, no investigational information about pharmaceutical agents that is outside US Food and Drug Administration–approved labeling has been presented in this article.
Financial disclosures: Dr Sakurai has received speakers honoraria from Yoshitomi Yakuhin and manuscript fee from Dainippon-Sumitomo Pharma within the past 3 years. Dr Suzuki has received manuscript or speakers fees from Astellas, Dainippon Sumitomo, Eli Lilly, Elsevier Japan, Janssen, Meiji Seika, Novartis, Otsuka, and Wiley Japan within the past 3 years. Dr Bies has received grants through National Institute of Mental Health (NIMH)/National Center for Theoretical Sciences (NCTS)/National Institute of Child Health and Human Development (NICHD)/National Institute of Allergy and Infectious Diseases (NIAID), Takeda, and Merck; has received travel support from Roche, Sanofi-Aventis, and Biogen Idec; has been a board member of Biogen Idec (clinical pharmacy advisory board) (paid), and a board member of International Society for Pharmacometrics (unpaid); has been a co–North American editor (paid) and executive editor for the British Journal of Clinical Pharmacology; and has served on editorial boards for Journal of Pharmacokinetics and Pharmacodynamics, Journal of Clinical Pharmacology, CPT:Pharmacometrics & Systems Pharmacology, and Biopharmaceutics and Drug Disposition. Dr Pollock receives research support from the National Institutes of Health, Canadian Institutes of Health Research, Brain Canada, and the Foundation of the Centre for Addiction and Mental Health; has been a member of the advisory board of Lundbeck Canada (final meeting was May 2009); has been a member of the advisory board of Forest Laboratories (final meeting was March 2008); has served one time as a consultant for Wyeth (October 2008) and Takeda (July 2007); and was a faculty member of the Lundbeck International Neuroscience Foundation (final meeting was April 2010). Dr Mimura has received grants and/or speakers honoraria from Asahi Kasei Pharma, Astellas, Daiichi Sankyo, Dainippon-Sumitomo, Eisai, Eli Lilly, GlaxoSmithKline, Janssen, Meiji-Seika, Mochida, MSD, Novartis, Otsuka, Pfizer, Shionogi, Takeda, Tanabe Mitsubishi, and Yoshitomi Yakuhin within the past 3 years. Dr Kapur has received grant support from Lundbeck and Roche; has served as a one-off consultant and/or speaker for AstraZeneca, Bristol-Meyers Squibb, Eli Lilly, Envivo, Janssen (Johnson & Johnson), Otsuka, Pfizer, and Takeda; serves on the scientific advisory boards for Lundbeck and Roche; and has received support from the National Institute for Health Research Mental Health Biomedical Research Centre at South London and Maudsley NHS Foundation Trust within the past 3 years. Dr Uchida has received grants from Astellas, Eisai, Otsuka, GlaxoSmithKline, Shionogi, Dainippon-Sumitomo, Eli Lilly, Mochida, Meiji-Seika, and Yoshitomi Yakuhin; has received speakers honoraria from Otsuka, Eli Lilly, Shionogi, GlaxoSmithKline, Yoshitomi Yakuhin, Dainippon-Sumitomo, Meiji-Seika, Abbvie, MSD, and Janssen within the past 3 years.
Funding/support: None.
Previous presentation: Some of the data were presented at the American Society of Clinical Psychopharmacology Annual Meeting; June 20–25, 2015; Miami, Florida.
Acknowledgments: The authors thank Yuya Mizuno, MD, and Kazunari Yoshida, MD, of Keio University School of Medicine, Tokyo, Japan, for clinical assessments, and Ai Ohtani of Keio University School of Medicine; Ken Kikuchi, MD, of Inokashira Hospital; and all staff at Inokashira Hospital, Tokyo, Japan, for their administrative support. Dr Mizuno has received manuscript fees or speakers honoraria from Sumitomo Dainippon, Astellas, and Eli Lilly; has received fellowship grants from the Japanese Society of Clinical Neuropsychopharmacology (Eli Lilly Fellowship for Clinical Psychopharmacology) and the Mochida Memorial Foundation for Medical and Pharmaceutical Research; and has received consultant fees from Bracket within the past 3 years. Dr Yoshida has received manuscript fees or speakers honoraria from Meiji Seika, Sumitomo Dainippon, and Eli Lilly within the past 3 years. Dr Kikuchi and Ms Ohtani have nothing to declare.
Supplementary material: Available at PSYCHIATRIST.COM.
REFERENCES
1. Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(7):1081–1090. PubMed doi:10.1016/j.pnpbp.2003.09.004
2. Argo TR, Crismon ML, Miller AL, et al. Texas Medication Algorithm Project Procedural Manual: Schizophrenia Treatment Algorithm. Austin, TX: Texas Department of State Health services; 2007.
3. Beasley CM Jr, Tollefson G, Tran P, et al. Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology. 1996;14(2):111–123. PubMed doi:10.1016/0893-133X(95)00069-P
4. Beasley CM Jr, Hamilton SH, Crawford AM, et al. Olanzapine versus haloperidol: acute phase results of the international double-blind olanzapine trial. Eur Neuropsychopharmacol. 1997;7(2):125–137. PubMed doi:10.1016/S0924-977X(96)00392-6
5. Chouinard G, Jones B, Remington G, et al. A Canadian multicenter placebo-controlled study of fixed doses of risperidone and haloperidol in the treatment of chronic schizophrenic patients. J Clin Psychopharmacol. 1993;13(1):25–40. PubMed doi:10.1097/00004714-199302000-00004
6. Kinon BJ, Kane JM, Johns C, et al. Treatment of neuroleptic-resistant schizophrenic relapse. Psychopharmacol Bull. 1993;29(2):309–314. PubMed
7. McEvoy JP, Hogarty GE, Steingard S. Optimal dose of neuroleptic in acute schizophrenia: a controlled study of the neuroleptic threshold and higher haloperidol dose. Arch Gen Psychiatry. 1991;48(8):739–745. PubMed doi:10.1001/archpsyc.1991.01810320063009
8. Lindenmayer JP, Citrome L, Khan A, et al. A randomized, double-blind, parallel-group, fixed-dose, clinical trial of quetiapine at 600 versus 1200 mg/d for patients with treatment-resistant schizophrenia or schizoaffective disorder. J Clin Psychopharmacol. 2011;31(2):160–168. PubMed doi:10.1097/JCP.0b013e31820f4fe0
9. Honer WG, MacEwan GW, Gendron A, et al; STACK Study Group. A randomized, double-blind, placebo-controlled study of the safety and tolerability of high-dose quetiapine in patients with persistent symptoms of schizophrenia or schizoaffective disorder. J Clin Psychiatry. 2012;73(1):13–20. PubMed doi:10.4088/JCP.10m06194
10. Goff DC, McEvoy JP, Citrome L, et al. High-dose oral ziprasidone versus conventional dosing in schizophrenia patients with residual symptoms: the ZEBRAS study. J Clin Psychopharmacol. 2013;33(4):485–490. PubMed doi:10.1097/JCP.0b013e3182977308
11. Dold M, Fugger G, Aigner M, et al. Dose escalation of antipsychotic drugs in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Res. 2015;166(1–3):187–193. PubMed doi:10.1016/j.schres.2015.04.024
12. Ng W, Uchida H, Ismail Z, et al. Clozapine exposure and the impact of smoking and gender: a population pharmacokinetic study. Ther Drug Monit. 2009;31(3):360–366. PubMed doi:10.1097/FTD.0b013e31819c7037
13. Uchida H, Suzuki T, Takeuchi H, et al. Low dose vs standard dose of antipsychotics for relapse prevention in schizophrenia: meta-analysis. Schizophr Bull. 2011;37(4):788–799. PubMed doi:10.1093/schbul/sbp149
14. World Health Organization. The ICD-10 Classification of Mental and Behavioural Disorders: Clinical Descriptions and Diagnostic Guidelines. Geneva, Switzerland: WHO; 1992.
15. Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261–276. PubMed doi:10.1093/schbul/13.2.261
16. Guy W. ECDEU Assessment Manual for Psychopharmacology, Revised. US Dept Health, Education, and Welfare publication (ADM) 76-338. Rockville, MD: National Institute of Mental Health; 1976:218–222.
17. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition. Washington DC: American Psychiatric Association; 1994.
18. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand suppl. 1970;212:11–19. PubMed doi:10.1111/j.1600-0447.1970.tb02066.x
19. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154:672–676. PubMed doi:10.1192/bjp.154.5.672
20. Sheiner LB, Rosenberg B, Marathe VV. Estimation of population characteristics of pharmacokinetic parameters from routine clinical data. J Pharmacokinet Biopharm. 1977;5(5):445–479. PubMed doi:10.1007/BF01061728
21. Beal B, Sheiner L. NONMEM User’s Guide, Part I. San Francisco, CA: University of California; 1992.
22. Uchida H, Mamo DC, Pollock BG, et al. Predicting plasma concentration of risperidone associated with dosage change: a population pharmacokinetic study. Ther Drug Monit. 2012;34(2):182–187. PubMed doi:10.1097/FTD.0b013e3182489a6f
23. Tsuboi T, Bies RR, Suzuki T, et al. Predicting plasma olanzapine concentration following a change in dosage: a population pharmacokinetic study. Pharmacopsychiatry. 2015;48(7):286–291. PubMed doi:10.1055/s-0035-1565070
24. Bigos KL, Pollock BG, Coley KC, et al. Sex, race, and smoking impact olanzapine exposure. J Clin Pharmacol. 2008;48(2):157–165. PubMed doi:10.1177/0091270007310385
25. Feng Y, Pollock BG, Coley K, et al. Population pharmacokinetic analysis for risperidone using highly sparse sampling measurements from the CATIE study. Br J Clin Pharmacol. 2008;66(5):629–639. PubMed
26. Hiemke C, Baumann P, Bergemann N, et al. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry. 2011;44(6):195–235. PubMed doi:10.1055/s-0031-1286287
27. Kapur S, Zipursky RB, Remington G, et al. 5-HT2 and D2 receptor occupancy of olanzapine in schizophrenia: a PET investigation. Am J Psychiatry. 1998;155(7):921–928. PubMed doi:10.1176/ajp.155.7.921
28. Nyberg S, Eriksson B, Oxenstierna G, et al. Suggested minimal effective dose of risperidone based on PET-measured D2 and 5-HT2A receptor occupancy in schizophrenic patients. Am J Psychiatry. 1999;156(6):869–875. PubMed doi:10.1176/ajp.156.6.869
29. Pajonk FG, Holzbach R, Naber D. Comparing the efficacy of atypical antipsychotics in open uncontrolled versus double-blind controlled trials in schizophrenia. Psychopharmacology (Berl). 2002;162(1):29–36. PubMed doi:10.1007/s00213-002-1055-9
30. Glick ID, Mankoski R, Eudicone JM, et al. The efficacy, safety, and tolerability of aripiprazole for the treatment of schizoaffective disorder: results from a pooled analysis of a sub-population of subjects from two randomized, double-blind, placebo-controlled, pivotal trials. J Affect Disord. 2009;115(1–2):18–26. PubMed doi:10.1016/j.jad.2008.12.017
31. Carpenter WT Jr, Heinrichs DW, Wagman AM. Deficit and nondeficit forms of schizophrenia: the concept. Am J Psychiatry. 1988;145(5):578–583. PubMed doi:10.1176/ajp.145.5.578
32. Voruganti L, Awad AG. Neuroleptic dysphoria: towards a new synthesis. Psychopharmacology (Berl). 2004;171(2):121–132. PubMed doi:10.1007/s00213-003-1648-y
33. Lecrubier Y, Quintin P, Bouhassira M, et al. The treatment of negative symptoms and deficit states of chronic schizophrenia: olanzapine compared to amisulpride and placebo in a 6-month double-blind controlled clinical trial. Acta Psychiatr Scand. 2006;114(5):319–327. PubMed doi:10.1111/j.1600-0447.2006.00887.x
34. Sakurai H, Bies RR, Stroup ST, et al. Dopamine D2 receptor occupancy and cognition in schizophrenia: analysis of the CATIE data. Schizophr Bull. 2013;39(3):564–574. PubMed doi:10.1093/schbul/sbr189
35. Agid O, Seeman P, Kapur S. The “delayed onset” of antipsychotic action—an idea whose time has come and gone. J Psychiatry Neurosci. 2006;31(2):93–100. PubMed


This PDF is free for all visitors!

