ABSTRACT
Objective: To evaluate the efficacy and safety of amphetamine extended-release tablets (AMPH ER TAB) in adults with attention-deficit/hyperactivity disorder (ADHD).
Methods: In a 5-week forced-dose titration phase, subjects were randomized to either oral double-blind AMPH ER TAB 5-mg starting dose or matching placebo, once daily in the morning. Safety and efficacy assessments were completed weekly. After visit 3, subjects received 20 mg for 14 ± 3 days before visit 5. At visit 5, efficacy assessments included the administration of serial Permanent Product Measure of Performance (PERMP) tests predose and at 0.5, 1, 2, 4, 8, 10, 12, 13, and 14 hours postdose. The primary efficacy endpoint was the mean PERMP Total score (PERMP-T) across postdose time points during the visit 5 serial PERMPs. Safety was monitored by adverse events (AEs) assessed at each visit, Columbia Suicide Severity Rating Scale (C-SSRS), vital signs, weight, physical examination, and assessment of sleep, appetite, mood, and psychotic AEs. The study was conducted from February 2019 to October 2019.
Results: Of 130 randomized subjects, 127 were in the intent-to-treat (ITT) population and 91 completed the study. The mean PERMP-T across all postdose time points at visit 5 was statistically significantly higher in the AMPH ER TAB group than in the placebo group (302.8 vs 279.6; P = .0043). Numerical differences favoring AMPH ER TAB were seen at all time points, with statistically significant improvements in the AMPH ER TAB group at 30 minutes and 1, 2, 4, 8, and 13 hours postdose, although the 10-, 12-, and 14-hour time points were not significant. Common AEs included decreased appetite, insomnia, and dry mouth. The majority of treatment-emergent AEs were mild to moderate in severity, and no serious AEs, as defined by the US Food and Drug Administration, were reported.
Conclusions: AMPH ER TAB demonstrated efficacy in treatment of symptoms of ADHD in adults, with an anticipated safety profile.
Trial Registration: ClinicalTrials.gov identifier: NCT03834766
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
J Clin Psychiatry 2022;83(5):22m14438
To cite: Cutler AJ, Childress AC, Pardo A, et al. Randomized, double-blind, placebo-controlled, fixed-dose study to evaluate the efficacy and safety of amphetamine extended-release tablets in adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2022;83(5):22m14438.
To share: https://doi.org/10.4088/JCP.22m14438
© 2022 Physicians Postgraduate Press, Inc.
aNeuroscience Education Institute, Lakewood Ranch, Florida
bDepartment of Psychiatry, SUNY Upstate Medical University, Syracuse, New York
cCenter for Psychiatry and Behavioral Medicine, Inc., Las Vegas, Nevada
dTris Pharma, Inc., Monmouth Junction, New Jersey
ePharmacoMetrica, La Fouillade, France
*Corresponding author: Stéphanie Duhoux, PhD, Tris Pharma, Inc., Medical Affairs, 2033 US 130, Monmouth Junction, NJ 08852 ([email protected]).
Attention-deficit/hyperactivity disorder (ADHD) typically manifests in childhood and has an estimated prevalence of 4.4.% in adults aged 18 to 44 years.1 Evidence exists that ADHD continues as a disorder meeting full diagnostic criteria in about 15% of individuals by age 25 years, but interfering symptoms persist in approximately 65%.2,3 Recent Multimodal Treatment Study of ADHD (MTA)4 findings establish ADHD as a chronic, waxing and waning disorder with periods of full remission that are more often temporary than sustained. ADHD in adults has been associated with deficits in educational and occupational attainment, social interacting, and self-esteem as well as obesity, service utilization, substance use,5–9 increased accident frequencies (eg, driving, operating heavy machinery), and increased morbidity and mortality, including higher suicide rates.10–12 Evidence suggests treatment can improve long-term outcomes, including decreasing suicidal behavior and mortality.9,11,13,14
Psychostimulants, especially amphetamines, are often prescribed for ADHD in adults.15 Specific attributes of a medication to meet individual needs in adults with ADHD include onset of efficacy early in the day that is maintained into the early evening hours,16 with once-daily dosing.17
Amphetamine extended-release tablet (AMPH ER TAB; Tris Pharma, Inc.; Monmouth Junction, New Jersey) is an amphetamine base product, 3.2:1 mixture of d– and l-amphetamine, and was developed using the proprietary LiquiXR drug delivery technology. LiquiXR technology enables a targeted pharmacokinetic profile that features a rapid onset of action followed by a smooth, continuous, extended-release profile allowing for once-daily dosing.18,19 This technology makes AMPH ER TAB the first continuous-release scalemic amphetamine formulation. Previous data19 showed that 18.8 mg of amphetamine extended-release oral suspension (AMPH EROS), an amphetamine base product, is bioequivalent to a single dose of 30 mg of mixed amphetamine salts.
The primary objective of this study was to evaluate the efficacy, safety, and tolerability of AMPH ER TAB compared with placebo for the treatment of ADHD in adults aged 18 to 60 years.
METHODS
Subjects
This study, performed from February 2019 to October 2019, enrolled subjects aged 18 to 60 years with a diagnosis of ADHD confirmed by Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition20 criteria based on the Adult ADHD Clinical Diagnostic Scale (ACDS),21,22 normal-range IQ (based on clinical opinion of the investigator), baseline Adult ADHD Investigator Symptom Rating Scale (AISRS)23,24 total score ≥ 26, and baseline Clinical Global Impressions–Severity of Illness scale (CGI-S)25 score ≥ 4. Female subjects of childbearing potential were non-lactating and non-pregnant during the duration of the study. All subjects provided written informed consent prior to any study procedure.
Exclusion criteria included lifetime history of bipolar disorder or any psychotic disorder or current major depression, generalized anxiety disorder, obsessive-compulsive disorder, panic disorder, or posttraumatic stress disorder, as established by the Mini-International Neuropsychiatric Interview (MINI) 7.0.226; history of chronic medical illnesses including untreated thyroid disease, peripheral vasculopathy, structural cardiac disorders, serious cardiac conditions, serious arrhythmias, cardiomyopathy, and family history of sudden death; clinically significant findings in vital signs at screening or history of uncontrolled hypertension or a resting systolic blood pressure (SBP) > 140 mm Hg or diastolic blood pressure (DBP) > 90 mm Hg; recent history or presence of alcohol or substance use disorder; history or presence of significant renal or hepatic disease; clinically significant abnormal electrocardiogram (ECG) or cardiac findings on physical examination; use of atomoxetine, monoamine oxidase inhibitors, or tricyclic antidepressants within 14 days of the baseline visit; use of gastrointestinal acidifying agents or urinary acidifying agents within 3 days of baseline visit; use of stimulant medications within 1 week of baseline visit; use of prohibited drugs or agents from the screening visit through the end of the study; an answer of “yes” to questions 4 or 5 of the Suicidal Ideation section of the Columbia Suicide Severity Rating Scale (C-SSRS)27 within the last 2 years; taking an investigational drug within 30 days prior to screening; known history of allergy/hypersensitivity to amphetamine or any of the components of AMPH ER TAB; and history of lack of clinical response to amphetamine.
Overall Study Design
After screening and baseline evaluations were completed, eligible subjects were randomized, in a parallel-study design, to double-blind AMPH ER TAB or matching placebo tablet taken orally once daily beginning the day after the baseline visit (study scheme shown in Figure 1) and entered a 5-week double-blind dose-titration phase. Subjects were then titrated up from 5 mg AMPH ER TAB/placebo, in 5-mg increments per week, and received a final dose of 20 mg for 14 ± 3 days prior to visit 5. Subjects who could not tolerate the study drug were discontinued from the study.
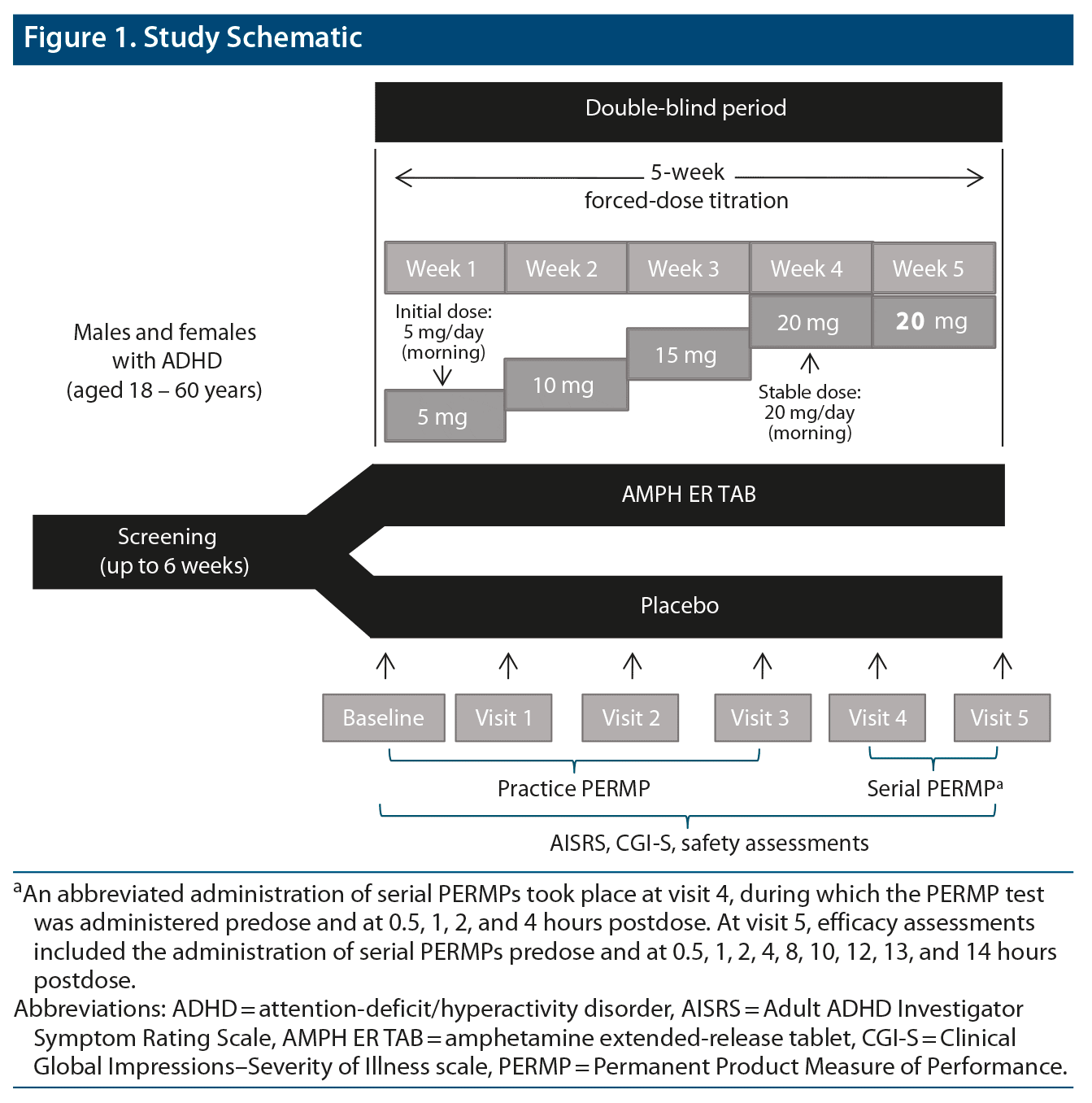
The primary efficacy endpoint was the comparison between AMPH ER TAB and placebo groups in the mean Permanent Product Measure of Performance Total scores (PERMP-Ts) averaged across all measured postdose time points at visit 5. Key secondary endpoints were treatment differences at each time point assessed (0.5, 1, 2, 4, 8, 10, 12, 13, and 14 hours postdose). Other key secondary objectives were to evaluate the effect of AMPH ER TAB compared with placebo on scores on the AISRS and CGI-S. Safety and tolerability were assessed by monitoring of adverse events (AEs) at each study visit, the C-SSRS, vital signs (blood pressure and heart rate), body weight, physical examination, and direct questioning about sleep quality, appetite, mood, and psychotic AEs.
The study (registered at ClinicalTrials.gov; identifier: NCT03834766) was conducted at 3 sites. The protocol, informed consent forms, and case report forms were reviewed and approved by an institutional review board (Copernicus Group IRB; Cary, North Carolina).
Treatments
Subjects were instructed to take double-blind AMPH ER TAB or placebo prior to 10:00 am, with or without food, by swallowing the tablet whole or by chewing it and swallowing. Study drug was administered onsite for visits 4 and 5. After visit 3, subjects received a final dose of 20 mg active product or placebo for 14 ± 3 days prior to visit 5.
Efficacy Measurements and Statistical Analysis
The PERMP is a validated, time-sensitive, skill-adjusted test consisting of simple math problems to be completed at multiple time points and provides the basis for an objective measure of the effect of treatment on the participant’s attention, productivity, and behavior (ability to initiate a task, self-monitor, stay on task, and complete written seatwork) in a simulated setting.28 PERMP-T is the sum of the number of math problems attempted plus the number of math problems answered correctly. PERMP-T ranges from 0 to 800 with higher scores indicating better performance. To scale for an individual’s math proficiency, a PERMP placement test was completed at screening or baseline. PERMP practice tests were also administered during baseline and visits 1 to 3. The CGI-S is a 7-point scale that is an overall assessment of the severity of illness at the time of assessment relative to the clinician’s experience with subjects of the same diagnosis.29 Possible ratings range from 1 (normal, not at all ill) to 7 (among the most extremely ill). The CGI-S was measured at screening, baseline, and study visits 1 through 5. The AISRS is a validated rating scale administered by trained raters to capture symptoms of ADHD in adult subjects. The scale has 18 items with adult prompts, based on the 18 diagnostic criteria symptoms of ADHD from DSM-5, each scored as follows: 0 (none), 1 (mild), 2 (moderate), 3 (severe). The maximum total score for the scale is 54 points.23,24 The AISRS was used to determine study eligibility and as an additional efficacy scale during visits 1 to 5.
The primary and key secondary efficacy analyses were performed on the modified intent-to-treat (mITT) population, defined as all randomized subjects who received at least one dose of study drug treatment and who had at least one PERMP-T score at study visit 5. Other secondary analyses were performed in the ITT population, defined as all randomized subjects who received at least 1 dose of study drug treatment, with treatment assignment based on the randomized treatment (regardless of actual treatment received). The safety population was defined as all enrolled subjects who received at least one dose of study drug.
The primary measure of intervention effect was the calculated treatment difference in mean PERMP-Ts averaged across all postdose time points between AMPH ER TAB and placebo. The PERMP-Ts at all postdose time points at study visit 5 were assessed using a linear mixed model repeated-measures (MMRM) analysis model using SAS PROC MIXED (SAS Institute; Cary, North Carolina) that included treatment, time, and treatment-by-time as fixed effects and participant as a random effect. The predose PERMP-T was included as a covariate. The difference between AMPH ER TAB and placebo was assessed at the α = .05 level of significance. Adjusted least-squares (LS) means were presented for each treatment across all postdose time points, and 95% confidence intervals (CIs) for the adjusted LS means were calculated. Treatment effect was estimated by the overall least square mean difference between the two treatment groups. The P value for the treatment comparison was also calculated. As an additional sensitivity to the primary analyses, the same MMRM model was run as specified previously in this paragraph including site as an additional factor. This was done to test if site had an impact on the primary outcome.
Safety Measurements
Safety was evaluated from reported AEs, changes in clinical laboratory values, changes in vital signs, ECG, C-SSRS, and physical examinations. Sleep, appetite, mood, and psychotic AEs were assessed by direct questioning as indicated in the ADHD Developing Stimulant Drugs for Treatment Draft Guidance.30 A treatment-emergent adverse event (TEAE) was defined as any event that was not present prior to the initiation of the drug treatment or any event already present that worsened in either intensity or frequency following exposure to the drug treatment AEs and TEAEs as well as vital signs, body weight, and occurrences of abnormal ECGs were assessed for relatedness and severity and summarized descriptively without any inferential statistical calculations. The C-SSRS scores were summarized by treatment.
RESULTS
Baseline Characteristics and Demography
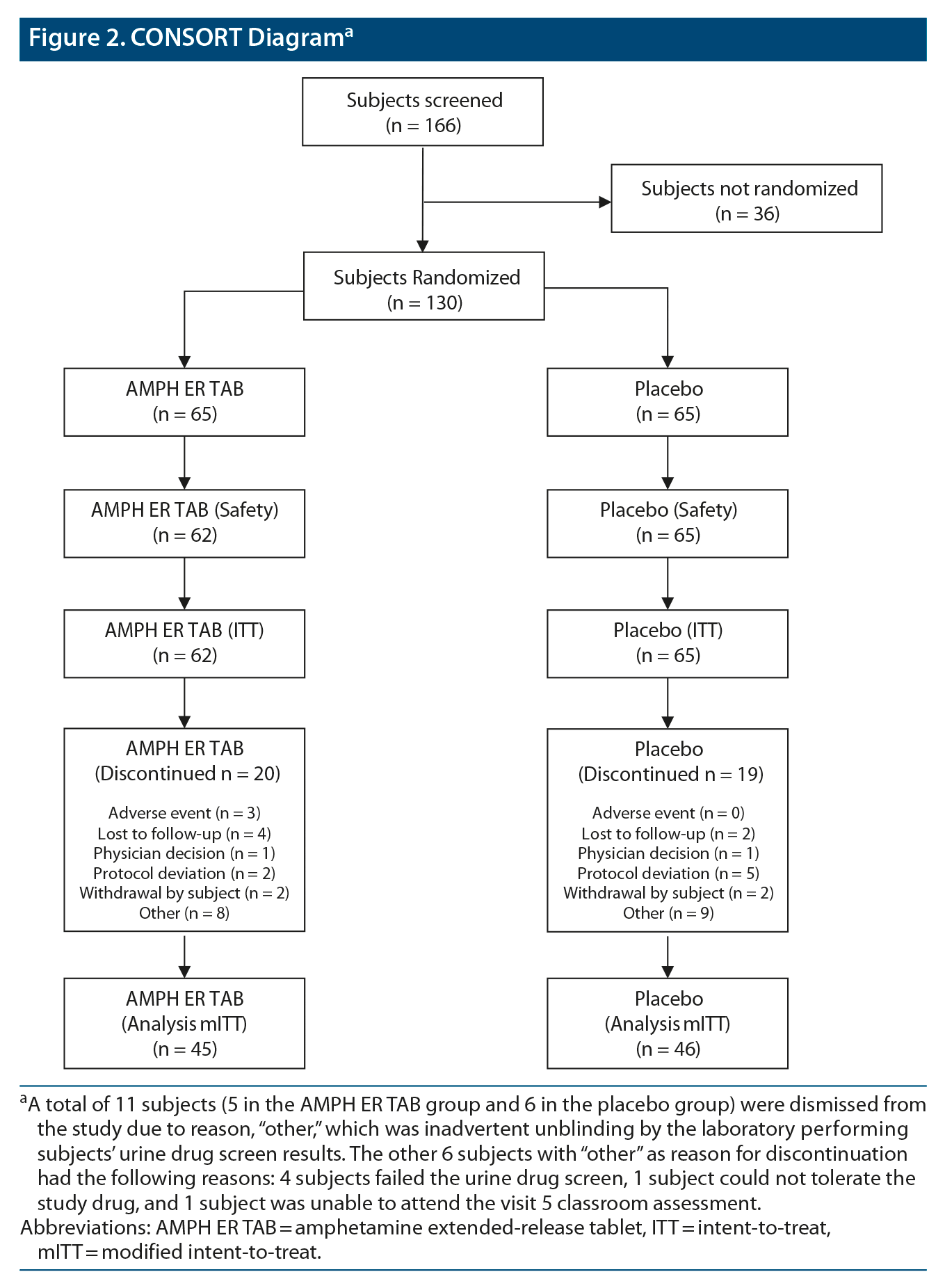
This study screened 166 subjects and randomized 130 equally to the two treatment arms. A breakdown of subject disposition, including reasons for nonrandomization, is provided in Figure 2.
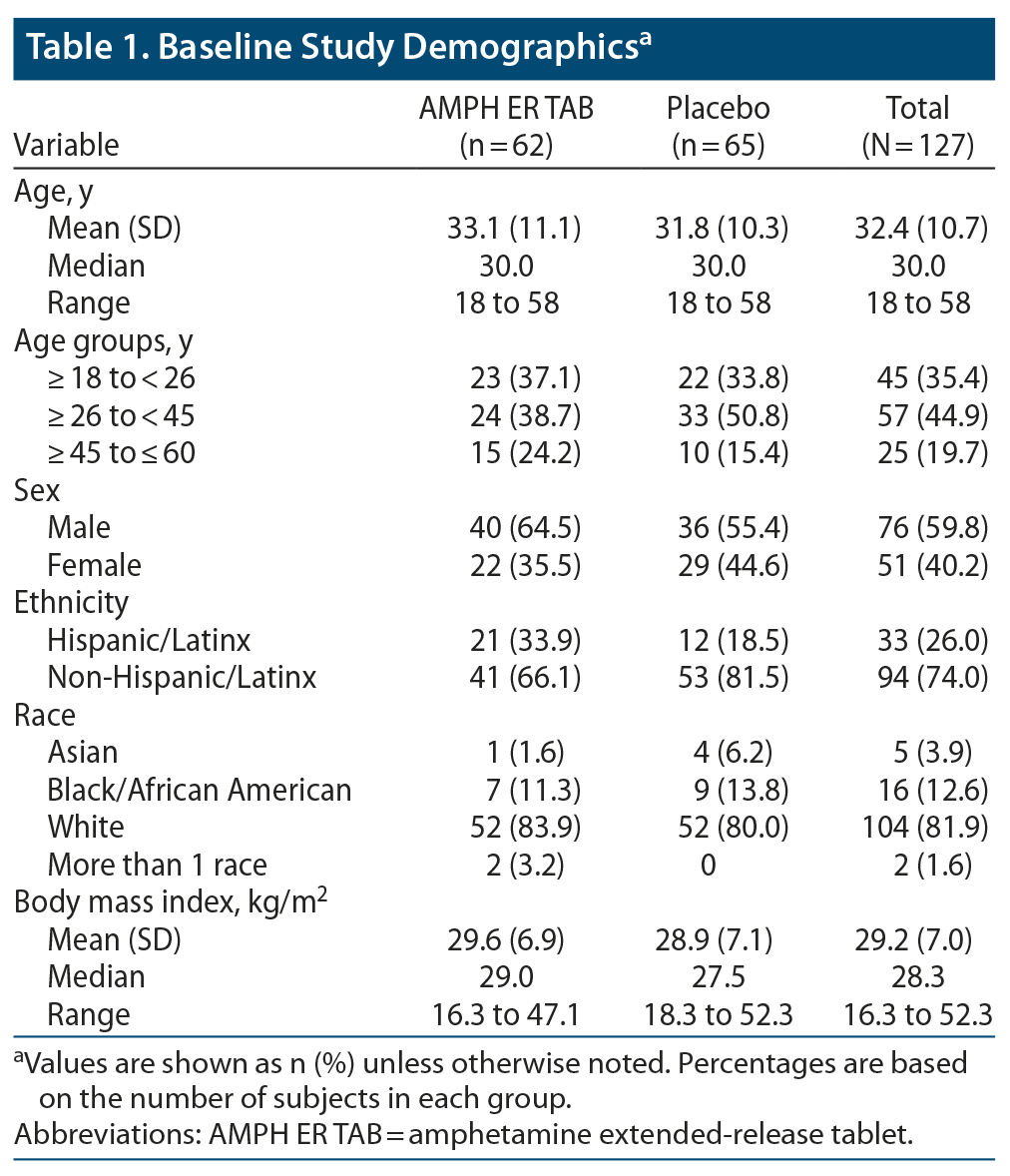
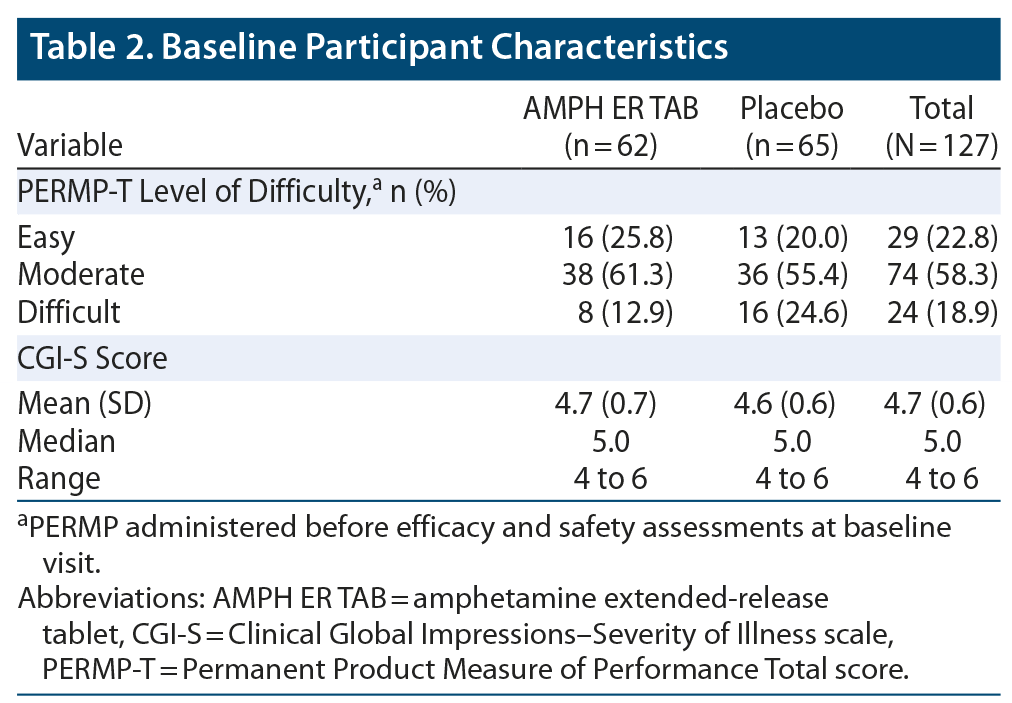
The two treatment groups were well balanced, with similar mean ages, body weights, body mass index (BMI), and height. About 80% of subjects were younger than 45 years of age. Baseline practice PERMP-Ts and assigned level of difficulty were similar between the two treatment groups. Demographic data are listed in Table 1, and baseline participant characteristics are summarized in Table 2.
Efficacy
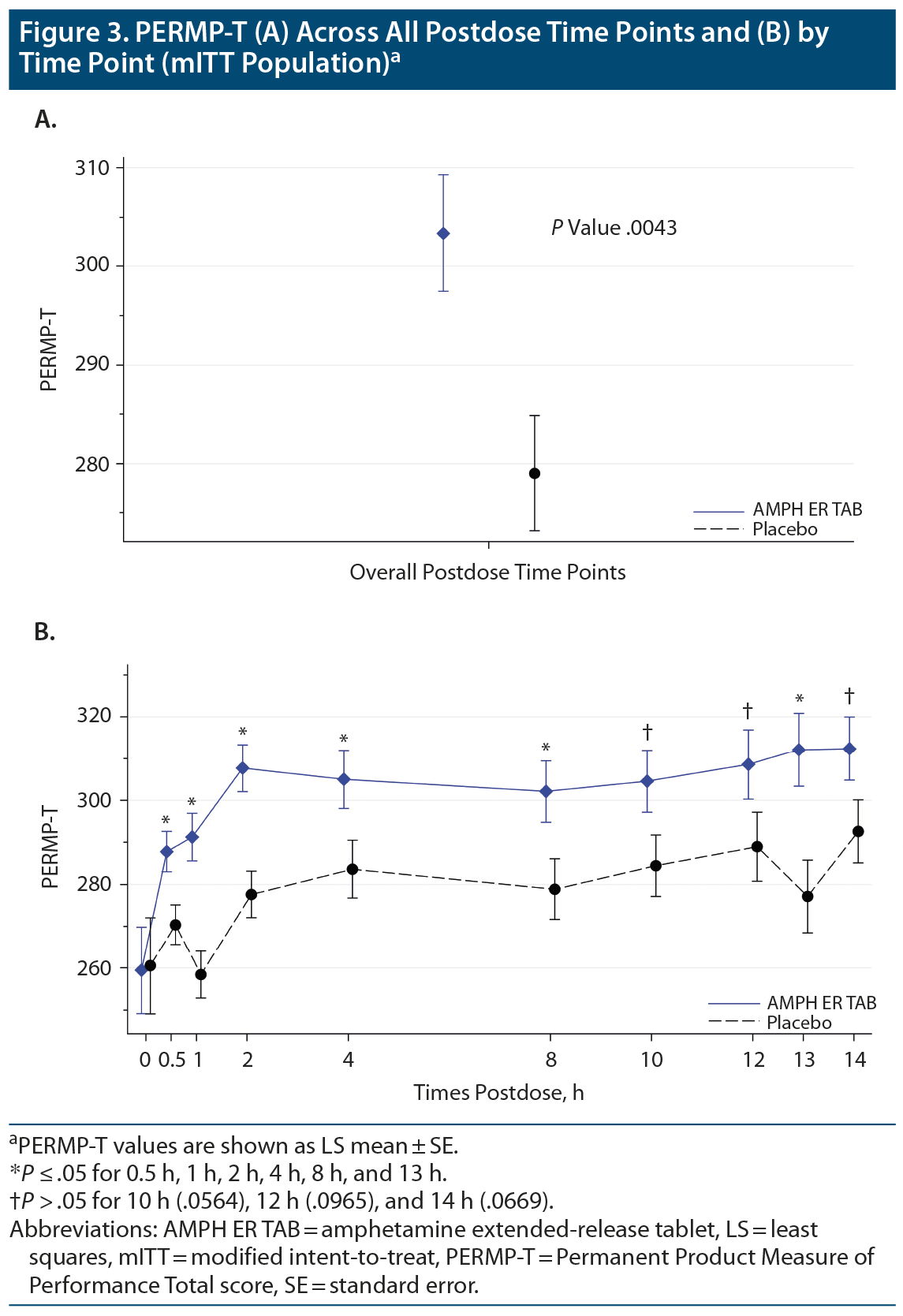
The mean predose PERMP-Ts at visit 5 (mITT population) were similar between the AMPH ER TAB group (259.5) and placebo (260.0). The primary endpoint was met: the LS mean postdose PERMP-T score was statistically significantly higher (improved) for the AMPH ER TAB treatment group (302.8) compared with placebo (279.6; P = .0043) (Figure 3A). No site effect was seen, as results were similar (P = .0048) when including site as an additional factor in the analysis. Results for the postdose treatment differences by time point are shown in Figure 3B. Based on the assessment, the first time point that was statistically significant was the 0.5-hour time point (P = .01 for the comparison between placebo and AMPH ER TAB treatment); and the last significant time point was noted at the 13-hour time point (P = .006). The treatment comparisons between placebo and AMPH ER TAB were statistically significantly improved at 1 hour (P < .001), 2 hours (P = .0003), 4 hours (P = .031), 8 hours (P = .026), and the aforementioned 0.5- and 13-hour postdose time points. The treatment differences were numerically better in those receiving AMPH ER TAB but not statistically significant at the 10-, 12-, and 14-hour time points.
The mean change from baseline in CGI-S score was consistently greater in the AMPH ER TAB group compared to the placebo group at each visit (Figure 4A), indicating a decrease in the severity of symptoms in the AMPH ER TAB group compared to the placebo group.
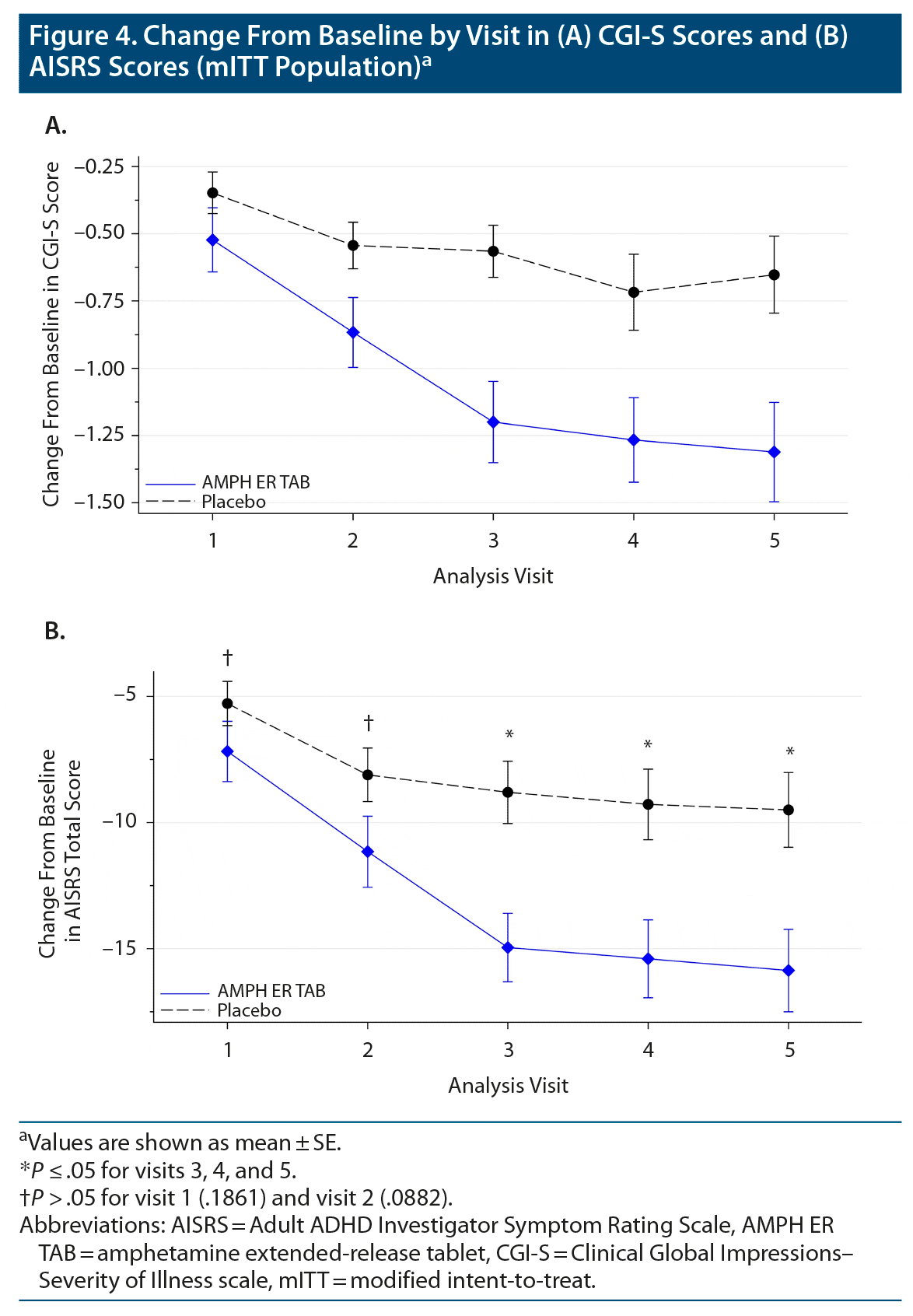
The other secondary endpoint was the change in AISRS score from baseline for subjects treated with AMPH ER TAB compared with those treated with placebo. The mean AISRS scores at baseline were similar for the active and placebo group (38.5 and 38.2, respectively). At each study visit, the mean ASIRS total scores decreased from baseline in both groups, but a statistically significantly greater reduction (improvement) was noted in the AMPH ER TAB group compared with placebo at visit 3 (P = .001), visit 4 (P = .004), and visit 5 (P = .005) (Figure 4B). The proportion of subjects who were treatment responders (defined as a subject who had a decrease from baseline of 50% or greater on the AISRS) in the AMPH ER TAB treatment group consistently increased from visit 1 (10 subjects, 16.1%) to visit 5 (23 subjects, 37.1%) and was proportionally greater than that observed in the placebo group (visit 3: 1 participant, 1.5%; and visit 5: 8 subjects, 12.3%).
Safety
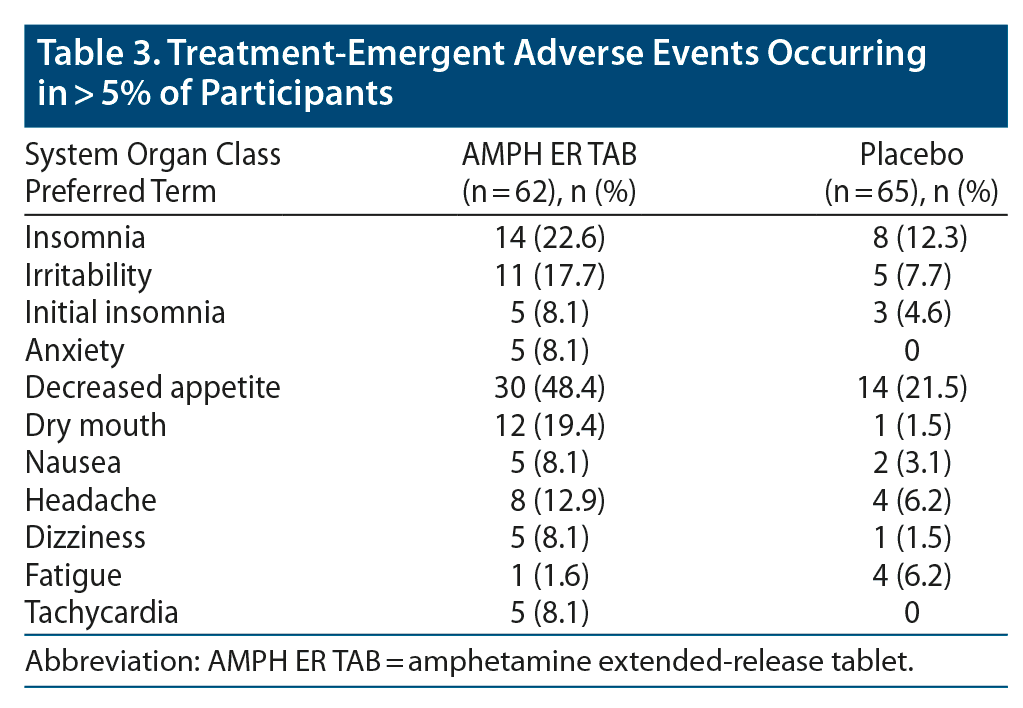
No deaths or serious AEs (as defined by the US Food and Drug Administration [FDA]) were reported in either treatment group in the study. Three subjects in the AMPH ER TAB group (4.8% of total) experienced AEs that led to discontinuation from the study; 1 experienced increased blood pressure, 1 experienced central nervous system stimulation, and 1 had anxiety; all 3 events were considered by the investigator to be probably related to study treatment and did not meet seriousness criteria as defined in the Code of Federal Regulations, 21CFR312.32.31 Greater proportions of subjects experienced AEs and TEAEs in the active treatment group compared with placebo (90% and 87%, respectively, for AMPH ER TAB and 60% and 54%, respectively, for placebo). Of those TEAEs, 86% were judged to be treatment-related in the AMPH ER TAB group compared with 48% for placebo. The most common TEAEs (reported at a frequency ≥ 5% and higher in the active treatment population compared with placebo) in the AMPH ER TAB group were decreased appetite (48.4%), insomnia (22.6%), dry mouth (19.4%), irritability (17.7%), headache (12.9%), and anxiety, nausea, dizziness, initial insomnia, and tachycardia (8.1% each) (Table 3). One participant in each treatment group experienced severe insomnia, and 1 subject (1.6%) in the AMPH ER TAB group experienced severe hypersensitivity (allergic reaction) possibly related to the study drug and was dismissed from the study.
Subjects receiving AMPH ER TAB experienced a small, non-clinically significant increase from baseline to visit 5 in mean ± SD SBP (116.8 ± 10.53 to 120.7 ± 10.94 mm Hg for AMPH ER TAB and 115.9 ± 10.84 to 114.5 ± 11.42 mm Hg for placebo) and DBP (74.1 ± 8.65 to 77.1 ± 8.53 mm Hg for AMPH ER TAB and 73.3 ± 9.24 to 71.6 ± 9.87 mm Hg for placebo). Increases were also observed for heart rate (73.0 ± 11.64 to 81.9 ± 10.40 bpm for AMPH ER TAB and 76.5 ± 10.71 to 74.8 ± 11.63 bpm for placebo). These hemodynamic findings are expected with stimulants such as amphetamine. All other vital sign measurements were similar between the two study groups. No clinically relevant differences between treatment groups were observed in the changes in mean ECG values from baseline to visit 5.
For the C-SSRS, at baseline 7 subjects (11.3%) in the AMPH ER TAB group and 7 subjects in the placebo group (10.8%) had any lifetime suicidal ideation or behavior; however, no subjects reported any suicidal ideation or behavior in the last 2 years or at any subsequent study visit. At visit 5, 9 subjects (14.5%) in the AMPH ER TAB group reported a change in sleeping upon query compared with 5 (7.7%) subjects in the placebo group. Subjects were also queried about changes in appetite; 8 (12.9%) of subjects in the AMPH ER TAB group reported a change in appetite since the last study visit compared with 2 (3.1%) in the placebo group. Finally, upon query about mood changes, 5 subjects (8.1%) in the AMPH ER TAB group reported a significant different mood since last visit compared with 1 participant (1.5%) in the placebo group. One participant in the AMPH ER TAB group reported feeling down, depressed, or hopeless, and 1 participant each in the AMPH ER TAB and placebo groups reported feeling more positive and cheerful and in a happier mood. Finally, 1 participant in the AMPH ER TAB group reported racing thoughts or feeling too much energy.
DISCUSSION
This study demonstrated a significant effect of AMPH ER TAB in adult patients aged 18 to 58 years with ADHD. The primary endpoint was met: the difference between AMPH ER TAB and placebo groups of the mean PERMP-T scores at visit 5 averaged across all time points was statistically significant, with a higher mean for subjects treated with AMPH ER TAB. Numerical differences favoring AMPH ER TAB were seen at all time points, with statistically significant improvements in PERMP-T for subjects in the AMPH ER TAB group at 30 minutes and 1, 2, 4, 8, and 13 hours postdose, although the differences at the 10-, 12-, and 14-hour time points were not significant. Subjects assigned to AMPH ER TAB displayed a consistently positive response throughout the duration of the study. On the other hand, the observed level of placebo response did not remain constant but fluctuated over time; this fluctuation may have contributed to the lack of statistically significant separation between AMPH ER TAB and placebo at 10, 12, and 14 hours. The efficacy profile of AMPH ER TAB was similar to the AMPH EROS efficacy data in 108 children aged 6 to 17 years, in which efficacy on the Swanson, Kotin, Agler, M-Flynn, and Pelham Rating Scale28,32 (SKAMP)–Combined scores was noted as early as 1 hour postdose (the earliest time point measured) through 13 hours postdose.33 An additional pilot study34 in children showed onset of clinical effect for AMPH EROS at 30 minutes postdose.
The purpose of AMPH ER TAB is to provide symptom relief for patients throughout the day, including early morning and into the evening. While short-acting medications might be an adequate symptom management tool for certain patients, guidelines recommend the use of long-acting stimulants,17,35 especially in adults, due to increased adherence and lower risk of abuse.36,37 Also, adults tend to prefer long-acting medications while also valuing speed of onset.16 A recent survey showed that when asked to make hypothetical ADHD treatment decisions, patients with ADHD favored a quick onset (24%) and long duration of action (42% of patients) while minimizing side effects (35%).16 This is in a context in which the most frequently prescribed long-acting stimulants can take up to 2 hours before showing an effect of symptoms,38 thus highlighting the need for multiple treatment options.
The pharmacokinetic profile of AMPH ER TAB was established to be bioequivalent to that of AMPH EROS, so that single doses of AMPH ER TAB 20 mg (swallowed whole or chewed) for both d– and l-amphetamine were determined to be bioequivalent to a 20-mg dose of the oral suspension (2.5 mg/mL) fasted and showed equivalent peak and overall exposure without apparent food effect.19 The LiquiXR drug delivery system allows for a rapid onset of effect followed by a smooth ascending concentration profile due to steady release with a profile indicative of once-daily dosing either in liquid or in tablet form.18 These results are reflected in the liquid formulation efficacy data in children33,34 and now in the tablet form in adults.
The safety profile of AMPH ER TAB, with common AEs being decreased appetite, insomnia, and dry mouth, was found to be comparable to those of other products within the stimulant class of ADHD medications, and the results of the AMPH ER TAB formulation were closely matched to those for AMPH EROS. In this study population of adults with ADHD, the AEs experienced by study subjects were expected for amphetamine products. There was an increase in blood pressure and heart rate, as has been seen in other studies of stimulants in adults with ADHD.39
There are a few limitations to this study. Driven by a change in FDA-recommended guidance, this study is the first of the efficacy and safety of a stimulant for treatment of ADHD symptoms in adults in which a double-blind, placebo-controlled fixed-dose design for the dose titration phase was used as opposed to an open-label flexible-dose titration phase occurring prior to the double-blind portion of an adult study measuring PERMP as the primary outcome measure throughout the day. Accordingly, it is difficult to contextually assess the impact of such a design change on the efficacy results, especially when comparing studies in which the older dose-optimization design was employed. The imposed fixed-dose design and the choice of a 20-mg dose might have impacted study outcomes, including hourly time point results, as some subjects may have benefited more from a lower or higher dose. This study included some variability in hourly time point results not seen in the oral suspension study, and while that may be indicative of some differences in the dose (forced vs optimized), in study populations (adults vs pediatric patients), primary outcome measure (PERMP vs SKAMP), or the differences in formulation, the durations of effect were the same as indicated by the statistically significant differences from placebo at the 13-hour postdose time point, and the efficacy noted in the study was reinforced by good response rates on both the AISRS and the CGI-S. This study was a placebo-comparator study, which does not allow for a head-to-head comparison with AMPH EROS or any other ADHD treatments, but the use of the PERMP-T as a primary efficacy variable is a common aspect of study designs for adult ADHD treatments. Other factors limiting the generalizability of this study are the duration of treatment of only 5 weeks, not allowing long-term assessments of efficacy and safety, and the characteristics of the sample that excluded most psychiatric comorbidities and other medical conditions. More than 50% of individuals with ADHD have at least one coexisting psychiatric disorder,40 and this study does not allow inference of the efficacy and safety of AMPH ER TAB in adults with ADHD and comorbidities.
Amphetamine has a proven track record as an efficacious treatment for improvement in ADHD symptomatology. The efficacy and safety of amphetamine, coupled with the rapid absorption and extended-release profile afforded by the LiquiXR drug delivery system demonstrated in this study, provide a new solid-dose treatment option that can be swallowed whole or chewed for patients with ADHD.
Submitted: February 25, 2022; accepted June 13, 2022.
Published online: July 20, 2022.
Relevant financial relationships: Dr Cutler is a consultant for AbbVie, Acadia, Alfasigma, Alkermes, Biogen, Boehringer Ingelheim, BlackThorn, Corium, Intra-Cellular Therapies, Ironshore, Janssen, Jazz, Karuna, Neumora, Neurocrine, Noven, Otsuka, Relmada, Sage Therapeutics, Sunovion, Supernus, Takeda, Teva, and Tris Pharma. He is a speaker for or has received promotional honoraria from AbbVie, Acadia, Alfasigma, Alkermes, Corium, Intra-Cellular Therapies, Ironshore, Janssen, Lundbeck, Neurocrine, Noven, Otsuka, Sunovion, Supernus, Takeda, Teva, and Tris Pharma. He is a member of the COMPASS Pathways Data Safety Monitoring Board (DSMB). Additionally, he is an employee and board member of the Neuroscience Education Institute. Dr Childress has received research support from Allergan, Takeda (Shire), Emalex, Akili, Otsuka, Purdue, Adlon, Sunovion, Tris Pharma, and Supernus. She in on the advisory board of Takeda (Shire), Noven, Otsuka, Sunovion, Tris Pharma, and Supernus. She is a consultant for Cingulate, Corium, Lumos, Otsuka, Purdue, Tris Pharma, KemPharm, and Supernus. She is a speaker for Takeda (Shire), Corium, Ironshore, Tris Pharma, and Supernus. She has received writing support from Takeda (Shire), Ironshore, Noven, Otsuka, Purdue, and Tris Pharma. Dr Gomeni is a consultant for Ironshore, Sunovion, Supernus, Teva, Biomedical Science Institutes, Nanomi BVs, Laboratorios Liconsa, Massachusetts General Hospital, UCB, Recordati Rare Diseases, Tris Pharma, F. Hoffmann-La Roche, 4SC AG, Autifony Therapeutics, and Indivior. Drs Pardo, Duhoux, Rafla, and Kando are employees of Tris Pharma. Mr King was an employee of Tris Pharma at the time the study was conducted. He now is an employee of, and hold stocks in, SCYNEXIS. Dr Kando owns stock in Johnson & Johnson and Takeda.
Funding/support: This article reports the results of a clinical study conducted by Tris Pharma, Inc., the developer and manufacturer of amphetamine extended-release tablets.
Role of the sponsor: Tris Pharma, Inc., was involved in all steps of the conduct and publication of the study.
Previous presentation: The results were originally reported at the 2021 American Professional Society of ADHD and Related Disorders, in Washington DC, on January 15, 2021. This study is registered on clinicaltrials.gov (identifier: NCT03834766). This manuscript is an original work and is not under consideration for publication anywhere other than The Journal of Clinical Psychiatry.
Acknowledgments: The authors greatly appreciate the support and assistance of Payal Naik, MPH, with the production of this manuscript. Payal Naik is an employee of Tris Pharma, Inc., Monmouth Junction, New Jersey.
CLINICAL POINTS
- There has been an increase in the diagnosis of attention-deficit/hyperactivity disorder (ADHD) in adults, and there is a need for data on treatment outcomes in the adult population.
- Amphetamine extended-release tablets (AMPH ER TAB) demonstrated efficacy in treatment of symptoms of ADHD in adults, with an anticipated safety profile.
- The efficacy profile of AMPH ER TAB coupled with its bioequivalence to amphetamine extended-release oral suspension (AMPH EROS) and its use of the same delivery technology, LiquiXR, comprising both immediate and extended-release components, places it as a new solid dose treatment option for adults with ADHD.
References (40)

- Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163(4):716–723. PubMed CrossRef
- Cherkasova M, Sulla EM, Dalena KL, et al. Developmental course of attention deficit hyperactivity disorder and its predictors. J Can Acad Child Adolesc Psychiatry. 2013;22(1):47–54. PubMed
- Faraone SV, Asherson P, Banaschewski T, et al. Attention-deficit/hyperactivity disorder. Nat Rev Dis Primers. 2015;1(1):15020. PubMed CrossRef
- Sibley MH, Arnold LE, Swanson JM, et al. Variable patterns of remission from ADHD in the Multimodal Treatment Study of ADHD. Am J Psychiatry. 2022;179(2):142–151. PubMed
- Barkley RA, Cunningham CE, Gordon M, et al. ADHD symptoms vs impairment: revisited. ADHD Rep. 2006;14(2):1–9. CrossRef
- Biederman J, Faraone SV, Spencer TJ, et al. Functional impairments in adults with self-reports of diagnosed ADHD: A controlled study of 1001 adults in the community. J Clin Psychiatry. 2006;67(4):524–540. PubMed CrossRef
- de Graaf R, Kessler RC, Fayyad J, et al. The prevalence and effects of adult attention-deficit/hyperactivity disorder (ADHD) on the performance of workers: results from the WHO World Mental Health Survey Initiative. Occup Environ Med. 2008;65(12):835–842. PubMed CrossRef
- Kessler RC, Adler L, Ames M, et al. The prevalence and effects of adult attention deficit/hyperactivity disorder on work performance in a nationally representative sample of workers. J Occup Environ Med. 2005;47(6):565–572. PubMed CrossRef
- Shaw M, Hodgkins P, Caci H, et al. A systematic review and analysis of long-term outcomes in attention deficit hyperactivity disorder: effects of treatment and non-treatment. BMC Med. 2012;10(1):99. PubMed CrossRef
- Barkley RA, Fischer M. Hyperactive child syndrome and estimated life expectancy at young adult follow-up: the role of ADHD persistence and other potential predictors. J Atten Disord. 2019;23(9):907–923. PubMed CrossRef
- Biederman J, DiSalvo M, Fried R, et al. Quantifying the protective effects of stimulants on functional outcomes in attention-deficit/hyperactivity disorder: a focus on number needed to treat statistic and sex effects. J Adolesc Health. 2019;65(6):784–789. PubMed CrossRef
- Furczyk K, Thome J. Adult ADHD and suicide. Atten Defic Hyperact Disord. 2014;6(3):153–158. PubMed CrossRef
- Boland H, DiSalvo M, Fried R, et al. A literature review and meta-analysis on the effects of ADHD medications on functional outcomes. J Psychiatr Res. 2020;123:21–30. PubMed CrossRef
- Chang Z, Quinn PD, O’Reilly L, et al. Medication for attention-deficit/hyperactivity disorder and risk for suicide attempts. Biol Psychiatry. 2020;88(6):452–458. PubMed CrossRef
- McGough JJ. Treatment controversies in adult ADHD. Am J Psychiatry. 2016;173(10):960–966. PubMed CrossRef
- Cambron-Mellott MJ, Mikl J, Matos JE, et al. Adult patient preferences for long-acting ADHD treatments: a discrete choice experiment. Patient Prefer Adherence. 2021;15:1061–1073. PubMed CrossRef
- CADDRA. Canadian ADHD Resource Alliance: Canadian ADHD Practice Guidelines. 4.1 Edition ed. CADDRA; 2020.
- Herman BK, King TR, Kando JC, et al. Novel, Modified-Release Drug Delivery Technology Containing Amphetamine Ion-Exchange Complexes. Presented at the American Professional Society for ADHD and Related Disorders Annual Meeting; January 18, 2019; Washington DC.
- Pardo A, Kando JC, King TR, et al. Single-dose pharmacokinetics of amphetamine extended-release tablets compared with amphetamine extended-release oral suspension. CNS Spectr. 2020;25(6):774–781. PubMed CrossRef
- American Psychiatric Association, ed. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. 2013.
- Adler L, Cohen J. Diagnosis and evaluation of adults with attention-deficit/hyperactivity disorder. Psychiatr Clin North Am. 2004;27(2):187–201. PubMed CrossRef
- Adler LA, Shaw DM, Kovacs K, et al. Diagnosing ADHD in children and adults. In: Adler LA, Spencer TJ, Wilens TE, eds. Attention Deficit Hyperactivity Disorder in Adults and Children. Cambridge: Cambridge University Press; 2015:16–23.
- Silverstein MJ, Faraone SV, Alperin S, et al. Validation of the expanded versions of the Adult ADHD Self-Report Scale v1.1 Symptom Checklist and the Adult ADHD Investigator Symptom Rating Scale. J Atten Disord. 2019;23(10):1101–1110. PubMed CrossRef
- Spencer TJ, Adler LA, Meihua Qiao, et al. Validation of the adult ADHD investigator symptom rating scale (AISRS). J Atten Disord. 2010;14(1):57–68. PubMed CrossRef
- Guy W. Clinical Global Impression. ECDEU Assessment Manual for Psychopharmacology-Revised. US Dept Health, Education and Welfare, ADAMHA. MIMH Psychopharmacology Research Branch; 1976:218–222.
- Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(suppl 20):22–33, quiz 34–57. PubMed
- Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. PubMed CrossRef
- Wigal SB, Wigal TL. The laboratory school protocol: its origin, use, and new applications. J Atten Disord. 2006;10(1):92–111. PubMed CrossRef
- Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont). 2007;4(7):28–37. PubMed
- Attention Deficit Hyperactivity Disorder: Developing Stimulant Drugs for Treatment Guidance for Industry. FDA Draft Guidance. FDA website. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/attention-deficit-hyperactivity-disorder-developing-stimulant-drugs-treatment-guidance-industry. 2019. Accessibility verified June 27, 2022.
- US Code of Federal Regulations - Title 21. National Archives. eCR website. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-312/subpart-B. Accessed May 10, 2022.
- Wigal SB, Gupta S, Guinta D, et al. Reliability and validity of the SKAMP rating scale in a laboratory school setting. Psychopharmacol Bull. 1998;34(1):47–53. PubMed
- Childress AC, Wigal SB, Brams MN, et al. Efficacy and safety of amphetamine extended-release oral suspension in children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2018;28(5):306–313. PubMed CrossRef
- Childress AC, Kando JC, King TR, et al. Early-onset efficacy and safety pilot study of amphetamine extended-release oral suspension in the treatment of children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2019;29(1):2–8. PubMed CrossRef
- Attention Deficit Hyperactivity Disorder: Diagnosis and Management. National Institute for Health and Care Excellence Guidelines. NICE website. https://www.nice.org.uk/guidance/ng87. 2018. Accessibility verified June 27, 2022.
- Bright GM. Abuse of medications employed for the treatment of ADHD: results from a large-scale community survey. Medscape J Med. 2008;10(5):111. PubMed
- Wilens TE, Adler LA, Adams J, et al. Misuse and diversion of stimulants prescribed for ADHD: a systematic review of the literature. J Am Acad Child Adolesc Psychiatry. 2008;47(1):21–31. PubMed CrossRef
- Childress A, Tran C. Current investigational drugs for the treatment of attention-deficit/hyperactivity disorder. Expert Opin Investig Drugs. 2016;25(4):463–474. PubMed CrossRef
- Wigal T, Brams M, Gasior M, et al; 316 Study Group. Randomized, double-blind, placebo-controlled, crossover study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder: novel findings using a simulated adult workplace environment design. Behav Brain Funct. 2010;6(1):34. PubMed CrossRef
- Sun S, Kuja-Halkola R, Faraone SV, et al. Association of psychiatric comorbidity with the risk of premature death among children and adults with attention-deficit/hyperactivity disorder. JAMA Psychiatry. 2019;76(11):1141–1149. PubMed CrossRef
This PDF is free for all visitors!




