
ABSTRACT
Objective: Delayed-release and extended-release methylphenidate (DR/ER-MPH), the first stimulant predicted to be absorbed primarily in the colon, demonstrated significant improvements in attention-deficit/hyperactivity disorder (ADHD) symptoms and functional impairment from awakening until evening versus placebo in clinical trials. The clinical significance of these improvements was explored post hoc by examining response and remission thresholds as well as safety in the context of dose optimization.
Methods: Data from the open-label, treatment-optimization phase of a phase 3 study of DR/ER-MPH in children (aged 6–12 years) with ADHD, as diagnosed by DSM-5 criteria and enrolled between July 2015 and March 2016, were analyzed. Thresholds for response (anchored to Clinical Global Impressions–Improvement scale [CGI-I] score of 1 or 2) and remission were applied to ADHD Rating Scale-IV (ADHD-RS-IV), Before School Functioning Questionnaire (BSFQ), and Parent Rating of Evening and Morning Behavior, Revised, Morning Subscale (PREMB-R AM) and Evening Subscale (PREMB-R PM) scores. Rates of response, remission, and treatment-emergent adverse events by starting dose were examined.
Results: Mean DR/ER-MPH dose increased from 29.7 mg/d at baseline (51% on 20 mg/d; 49% on 40 mg/d) to 66.2 mg/d at week 6. At week 6, most participants achieved response/remission thresholds (response/remission: ADHD-RS-IV: 97%/89%; BSFQ: 98%/94%; PREMB-R AM: 94%/98%; PREMB-R PM: 91%/84%). More participants starting on a 40-mg versus 20-mg dose achieved thresholds at week 1 (P < .02). Weekly treatment-emergent adverse event rates over the open-label period were similar between starting doses.
Conclusions: When DR/ER-MPH dosing was optimized for ADHD symptom control throughout the day, the majority of participants achieved thresholds indicating all-day control of ADHD symptoms and functional impairment to the level of their non-ADHD peers.
Trial Registration: Data used in this post hoc analysis came from the study with ClinicalTrials.gov identifier: NCT02493777
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
J Clin Psychiatry 2021;82(4):21m13914
To cite: Childress AC, Cutler AJ, Po MD, et al. Symptomatic and functional response and remission from the open-label treatment-optimization phase of a study with DR/ER-MPH in children with ADHD. J Clin Psychiatry. 2021;82(4):21m13914.
To share: https://doi.org/10.4088/JCP.21m13914
© Copyright 2021 Physicians Postgraduate Press, Inc.
aCenter for Psychiatry and Behavioral Medicine, Inc., Las Vegas, Nevada
bSUNY Upstate Medical University, Syracuse, New York
cHighland Therapeutics Inc., Toronto, Ontario, Canada
dIronshore Pharmaceuticals & Development, Inc., Camana Bay, Grand Cayman, Cayman Islands
eIronshore Pharmaceuticals Inc., Durham, North Carolina
*Corresponding author: Ann Childress, MD, Center for Psychiatry and Behavioral Medicine, 7351 Prairie Falcon Rd, Ste 160, Las Vegas, NV 89128 ([email protected]).
Recent guidelines for the treatment of attention-deficit/hyperactivity disorder (ADHD)1,2 emphasize the importance of treatments with durations of effect that extend beyond the school- and workday. These guidelines also stress the importance of implementing treatments that target functional impairment, commonly the primary cause for seeking treatment for ADHD,3 in addition to symptom control. Despite formulation improvements, all-day clinical efficacy with a single stimulant dose has remained an unmet need in adults4 and youth5,6 with ADHD.
HLD200, a delayed-release and extended-release formulation of methylphenidate (DR/ER-MPH; trade name: JORNAY PM) approved by the US Food and Drug Administration (FDA) for the treatment of ADHD in individuals aged 6 years and older,7 is the first stimulant that is predicted to be absorbed primarily in the colon following evening administration without an immediate-release component. Because the colon is a less efficient site of absorption compared to the upper gastrointestinal tract,8 colonic absorption is predicted to underlie several of the pharmacokinetic properties of DR/ER-MPH, including a gradual ascending curve in the early morning, attenuated peak plasma concentration, protracted elimination phase into the evening, and a dose-dependent duration of effect.9,10 In two phase 3 studies of children with ADHD,11,12 treatment with DR/ER-MPH improved symptoms as well as functional impairment during the early morning, over a laboratory classroom test day, and in the late afternoon/evening versus placebo.
Primary and secondary efficacy endpoints, including those from the two phase 3 studies of DR/ER-MPH, are typically averages of continuous measures, which summarize a range of individual treatment outcomes. A categorical outcome measure based on a threshold of treatment success (ie, proportion of patients that achieve the thresholds) may be more clinically intuitive than information from a continuous outcome measure based on group averages.13 As such, applying established cutoffs can help clinicians understand the clinical meaningfulness of an aggregate treatment effect and help predict positive individual patient outcomes.13,14 A number of thresholds using various symptom scales have been used to determine symptomatic response, including a decrease in ADHD Rating Scale-IV (ADHD-RS-IV) or Swanson, Nolan, and Pelham-IV-18 (SNAP-IV-18) total score of 25%–30% to define response or 40%–50% to define a more robust response, either alone or in combination with a Clinical Global Impressions–Improvement scale (CGI-I) score of 1 (very much improved) or 2 (much improved).15–24 Weiss and colleagues25 identified a decrease of 40% in ADHD-RS-IV total score as being most closely aligned with individuals being very much or much improved (CGI-I score = 1 or 2).
Some have proposed utilizing more robust thresholds to achieve scores within the range of unaffected individuals, which has been termed symptomatic or functional remission.26–28 In the context of ADHD, a neurodevelopmental disorder, remission is contextualized within the framework of currently available treatments and does not reference duration of improvement. Symptomatic remission in ADHD has commonly been operationalized as ADHD-RS-IV or SNAP-IV-18 scores of ≤ 18, indicating mean scores of ≤ 1 (“mild or less”) per item.13,26 This threshold for remission captured 88% of children in the control group of the National Institute of Mental Health Collaborative Multisite Multimodal Treatment Study of Children with Attention-Deficit/Hyperactivity Disorder (MTA).13
Recently, decreases in total score of at least 45%, 49%, and 29% were determined to be clinically meaningful (anchored to CGI-I score = 1 or 2 [very much or much improved]) for the Before School Functioning Questionnaire (BSFQ); Parent Rating of Evening and Morning Behavior, Revised, Morning Subscale (PREMB-R AM); and PREMB-R Evening Subscale (PREMB-R PM), respectively.29 For the BSFQ, PREMB-R AM, and PREMB-R PM, age-appropriate norm-referenced cutoffs30 have also been determined to identify severity (screening risk, mild, moderate, and severe) of functional impairment during the early morning and late afternoon/evening. Individuals with scores below screening risk (< 80th percentile) represent a level of functional impairment that is indistinguishable from the general population (ie, functional remission). Indeed, scores below the screening risk threshold on the BSFQ, PREMB-R AM, and PREMB-R PM captured 87%–89% of youth with no ADHD and no comorbidities.30
The goal of these post hoc analyses was to examine symptom and functional impairment scores by applying categorical cutoffs for response and remission in the context of DR/ER-MPH dosing during the 6-week, open-label, treatment-optimization period of a phase 3 study.12 Rates of treatment-emergent adverse events (TEAEs) in the context of DR/ER-MPH dosing were also examined.
METHODS
Participants
Children (aged 6–12 years) were enrolled in the study if they met the predefined entry criteria, as described in a previous report.12 Briefly, key inclusion criteria included diagnosis of ADHD based on the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition31 criteria and confirmed with the Mini-International Neuropsychiatric Interview for Children and Adolescents; baseline ADHD-RS-IV score ≥ 90th percentile normalized for sex and age in at least one of the following categories: inattentive, hyperactive-impulsive, or total score, and a total score of ≥ 26 at baseline; prior response to MPH treatment; and parent/guardian confirmation of before-school functional impairment and difficulties performing morning routine, with a weekday morning routine of at least 30 minutes.
Study Design
This pivotal phase 3, multicenter, laboratory classroom study (NCT02493777) of DR/ER-MPH in children with ADHD took place between July 2015 and March 2016 and was conducted in 3 distinct phases: (1) a screening/washout phase (up to 4 weeks, with washout of ADHD treatment for ≥ 5 days); (2) a 6-week, open-label, DR/ER-MPH treatment–optimization phase; and (3) a 1-week, double-blind, randomized, placebo-controlled, parallel-group phase concluding with a laboratory classroom test day. At baseline, all participants received DR/ER-MPH, either 20 mg or 40 mg, once daily at 8:00 pm (± 30 minutes), with the starting dose dependent on their previous treatment history. Over the 4 subsequent study visits (weeks 1, 2, 3, and 4), dose titrations were permitted in 20- or 40-mg increments or decrements until an optimal daily dose was achieved or a maximum daily dose of 100 mg/d or 3.7 mg/kg was reached. Adjustments to the evening administration time in 30- to 60-minute increments or decrements were permitted (between 6:30 pm and 9:30 pm).
Optimized dose and administration time were predefined as those that produced meaningful control during the morning and throughout the day while remaining safe and well tolerated with ≥ 33% improvement from baseline in ADHD-RS-IV, BSFQ, and Conners’ Global Index–Parent [CGI-P] scores. The final permitted dose and administration time adjustments were made at the week 4 visit, after which dose and administration time were to be maintained from the week 5 visit through to randomization at the week 6 visit (after completing the 6-week treatment-optimization phase).
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, and all participants and parents/legal guardians provided informed assent and consent, respectively, under procedures approved by each site’s Institutional Review Board.
Assessments and Statistical Analyses
Descriptions of assessments, including timing and scoring, are listed in Supplementary Table 1. Post hoc analyses were performed in the intent-to-treat (ITT) population, defined as randomized participants who received at least one dose of double-blind study drug and had at least one post-baseline evaluation of the primary efficacy variable. Dose, number of participants at each dose level, ADHD-RS-IV scores, and BSFQ scores were summarized at each time point using descriptive statistics; the PREMB-R AM and PREMB-R PM were administered only at baseline and week 6. Rating scale scores reflected symptoms or functional impairment over the preceding week. Percentage change from baseline for ADHD-RS-IV, BSFQ, and PREMB-R AM/PM (week 6 only) total scores was calculated as

The response thresholds applied in this study were determined using the same external anchor (CGI-I score = 1 or 2, indicating very much or much improved). The following response thresholds were used: ADHD-RS-IV score, ≥ 40% decrease from baseline; BSFQ score, ≥ 45% decrease from baseline, PREMB-R AM score, ≥ 49% decrease from baseline; and PREMB-R PM score, ≥ 29% decrease from baseline.25,29 Similarly, the remission thresholds that were applied were also chosen because they identify the majority of youth without ADHD (Supplementary Table 1).13,30 For children 6–8 and 9–12 years of age, respectively, the following remission thresholds were used: BSFQ, ≤24 and ≤ 21; PREMB-R AM, ≤4 and ≤ 3; and PREMB-R PM, ≤10 and ≤ 8.30 The proportion achieving each threshold was compared, using the Fisher exact test, between those who received a starting dose of 20 mg and those who received 40 mg. TEAEs were assessed by general query at weekly visits. TEAEs of special interest included those related to sleep and appetite. Unlike all other TEAEs, which were spontaneously reported, sleep-related TEAEs were directly queried by asking about onset, quality, and quantity of sleep. The number and percentage of participants reporting TEAEs and the number of TEAEs were summarized by baseline dose (ie, those starting on 20 mg vs 40 mg).
RESULTS
Participants
Participant disposition as well as demographics and baseline characteristics have been previously reported.12 Briefly, the ITT population (N = 117), in which all analyses were performed, was 68.4% male (80/117), mean ± SD age was 9.4 ± 1.63 years, and presentation was mostly combined type (86.3%) with some predominantly inattentive (13.7%). The mean ± SD baseline ADHD-RS-IV score was 42.5 ± 6.60. The mean ± SD baseline BSFQ score was 40.7 ± 10.28, which corresponds to meeting the 93rd percentile threshold or moderate severity of early morning functional impairment,30 and 5 (4.3%) of 117 participants had BSFQ scores below the 80th percentile or screening risk at baseline. The mean ± SD baseline PREMB-R AM score was 5.7 ± 2.44, which corresponds to meeting the 80th percentile threshold or screening risk,30 and 23 (19.7%) of 117 participants had PREMB-R AM scores below screening risk at baseline. The mean ± SD baseline PREMB-R PM score was 17.2 ± 4.17, which corresponds to meeting the 93rd percentile threshold or moderate severity of late afternoon/evening functional impairment,30 and 3 (2.6%) of 117 participants had PREMB-R PM scores below screening risk at baseline.
Dose Titration
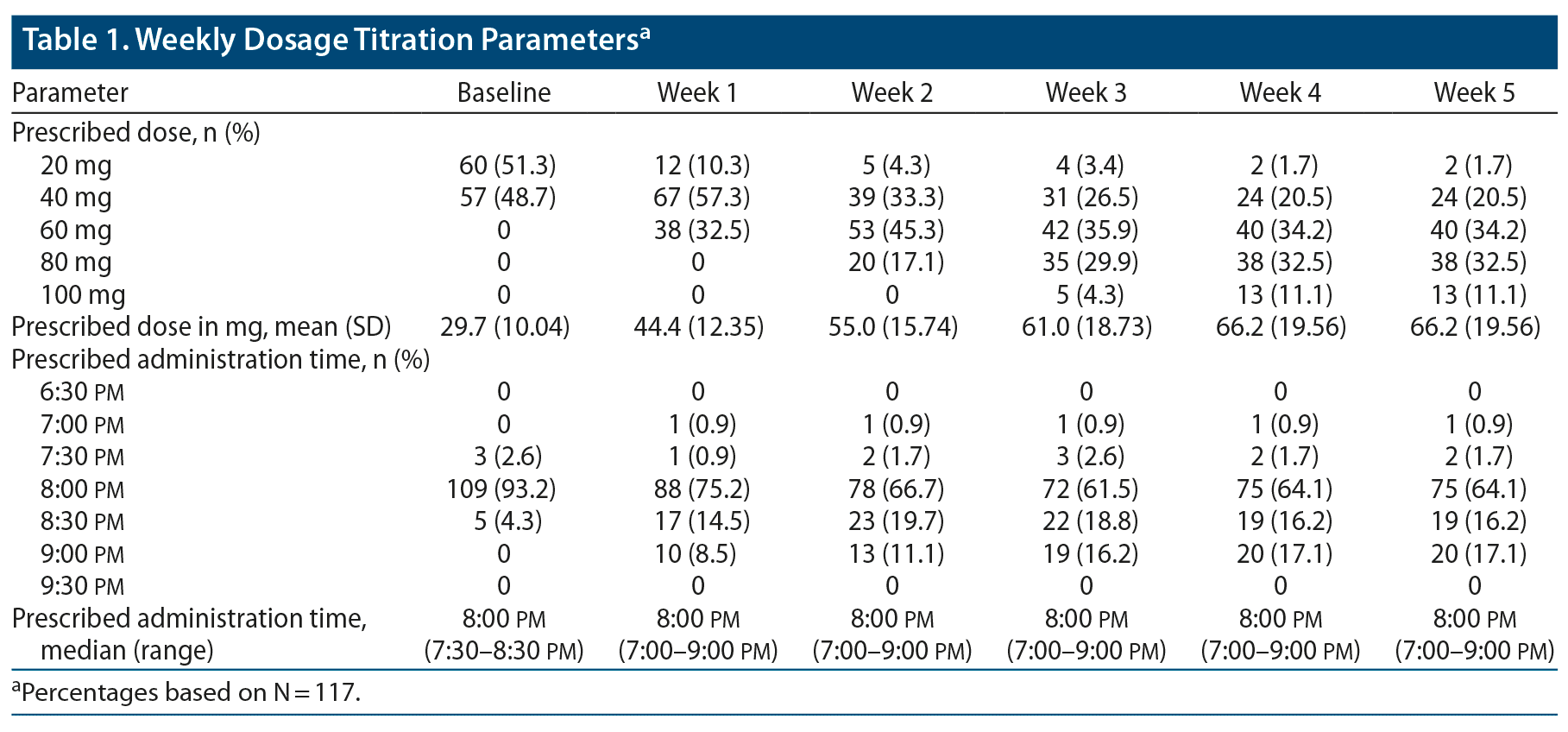
Investigators were provided a starting dose manual that prespecified DR/ER-MPH starting doses based on previous methylphenidate dosing. The starting doses were calculated based on available pharmacokinetic and comparative bioavailability data and rounded down to 20 mg or 40 mg of DR/ER-MPH. On the basis of previous dosing history, approximately half of participants (51.3%) received 20 mg and approximately half (48.7%) received 40 mg as their initial dose at baseline (Table 1). The final mean ± SD optimized dose was 66.2 ± 19.56 mg,12,36 with 87.2% optimized to 40-, 60-, or 80-mg doses (Table 1). At the beginning of the open-label period, the majority of participants (93.2%) had a prescribed administration time of 8:00 pm. At the end of the open-label period, the most common (64.1%) administration time was still 8:00 pm, with most adjustments shifting to administration later in the evening (Table 1).
Substantial Mean Improvements in ADHD Symptoms and Functional Impairment During the Early Morning and Late Afternoon/Evening
ADHD-RS-IV (Supplementary Figure 1A) and BSFQ (Supplementary Figure 1B) mean scores improved markedly from the baseline assessment (reflecting the previous untreated week) to the week-1 assessment (reflecting the first week of open-label treatment). The randomization criterion of ≥ 33% improvement on the ADHD-RS-IV and BSFQ was achieved after 1 week of DR/ER-MPH treatment (Supplementary Figure 1); therefore, dose increases over the subsequent weeks (Table 1) were guided by ADHD-RS-IV and BSFQ scores (but not by a specific target) and clinical judgement to optimize treatment effect throughout the day. Following the first week of treatment, ADHD-RS-IV and BSFQ mean scores continued to improve week-over-week during the treatment-optimization period, generally mirroring increases in mean doses (Table 1). PREMB-R AM and PREMB-R PM scores also improved substantially from baseline to week 6 (not measured in intervening weeks): mean ± SD PREMB-R AM scores improved from 5.7 ± 2.44 at baseline to 0.8 ± 1.11 at week 6, and PREMB-R PM scores improved from 17.2 ± 4.17 at baseline to 5.3 ± 4.20 at week 6.
Achievement of Response and Remission Thresholds After the 6-Week, Open-Label, DR/ER-MPH Treatment-Optimization Phase
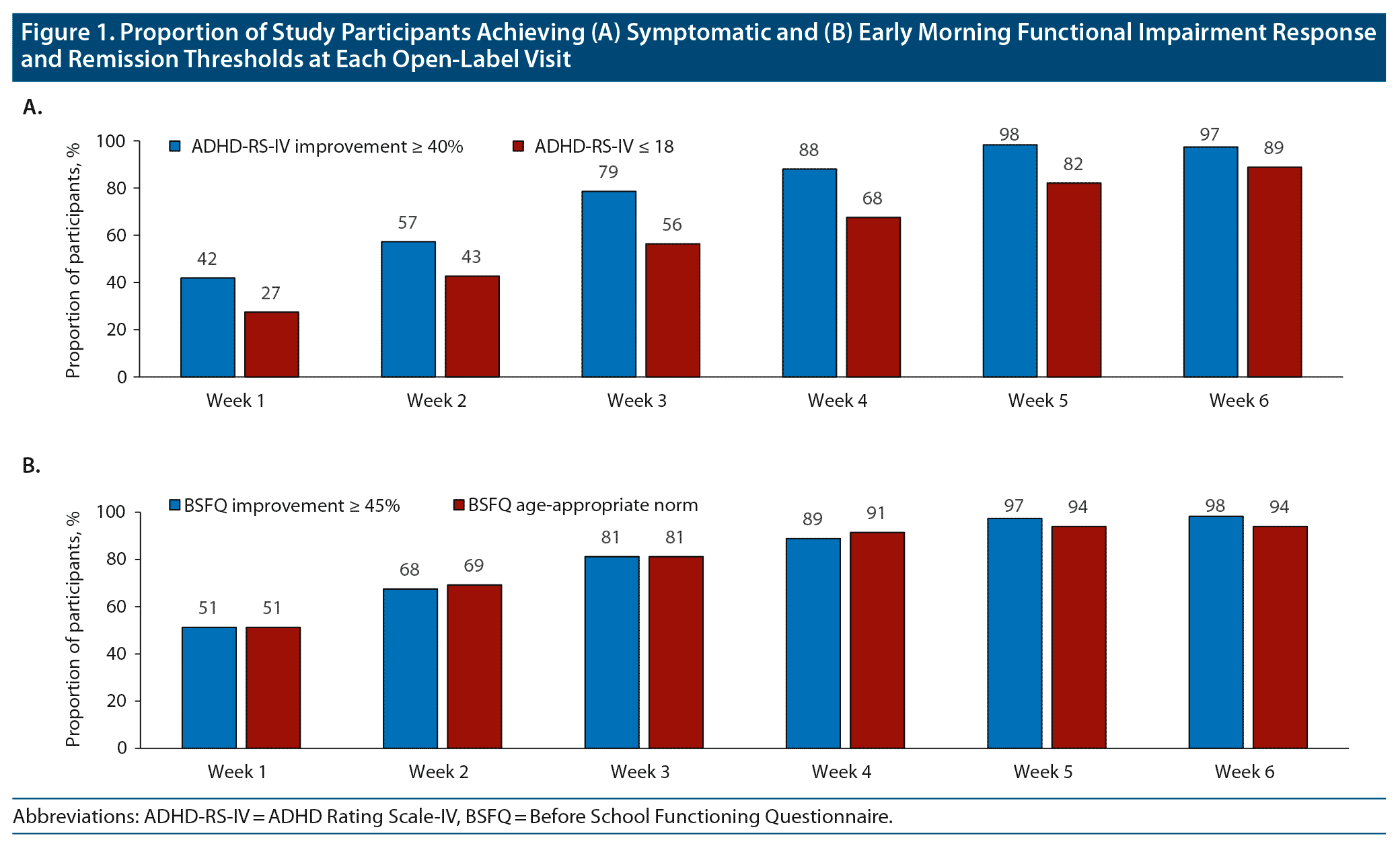
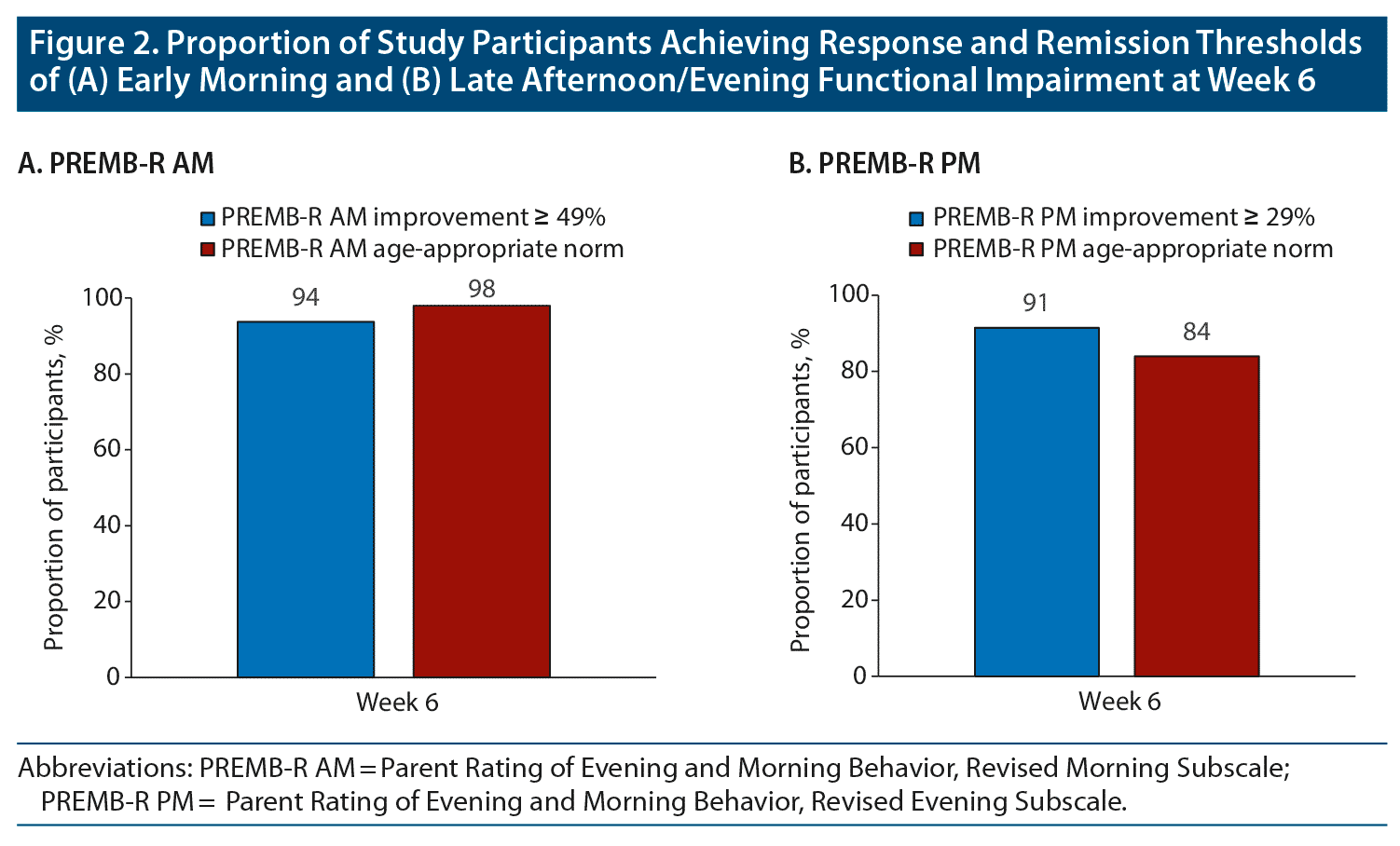
Achievement of response and remission thresholds increased from the beginning to the end of the treatment-optimization period. After 1 week of treatment with open-label DR/ER-MPH, 42% of participants achieved symptomatic response and 27% achieved symptomatic remission by ADHD-RS-IV thresholds (Figure 1A). After 6 weeks of treatment with open-label DR/ER-MPH, 97% of participants achieved symptomatic response and 89% achieved symptomatic remission by ADHD-RS-IV thresholds (Figure 1A). After 1 week of treatment with open-label DR/ER-MPH, 51% of participants achieved response and 51% achieved remission in early morning functional impairment by BSFQ thresholds (Figure 1B). After 6 weeks of treatment with open-label DR/ER-MPH, 98% and 94% of participants achieved response and remission, respectively, in early morning functional impairment based on the BSFQ (Figure 1B). Similarly, 94% and 98% achieved response and remission, respectively, in early morning functional impairment based on the PREMB-R AM (Figure 2A). After 6 weeks of treatment with open-label DR/ER-MPH, 91% and 84% of participants achieved response and remission, respectively, in late afternoon/evening functional impairment based on PREMB-R PM thresholds (Figure 2B).
Improvement in ADHD Symptoms and Early Morning Functional Impairment by Starting Dose
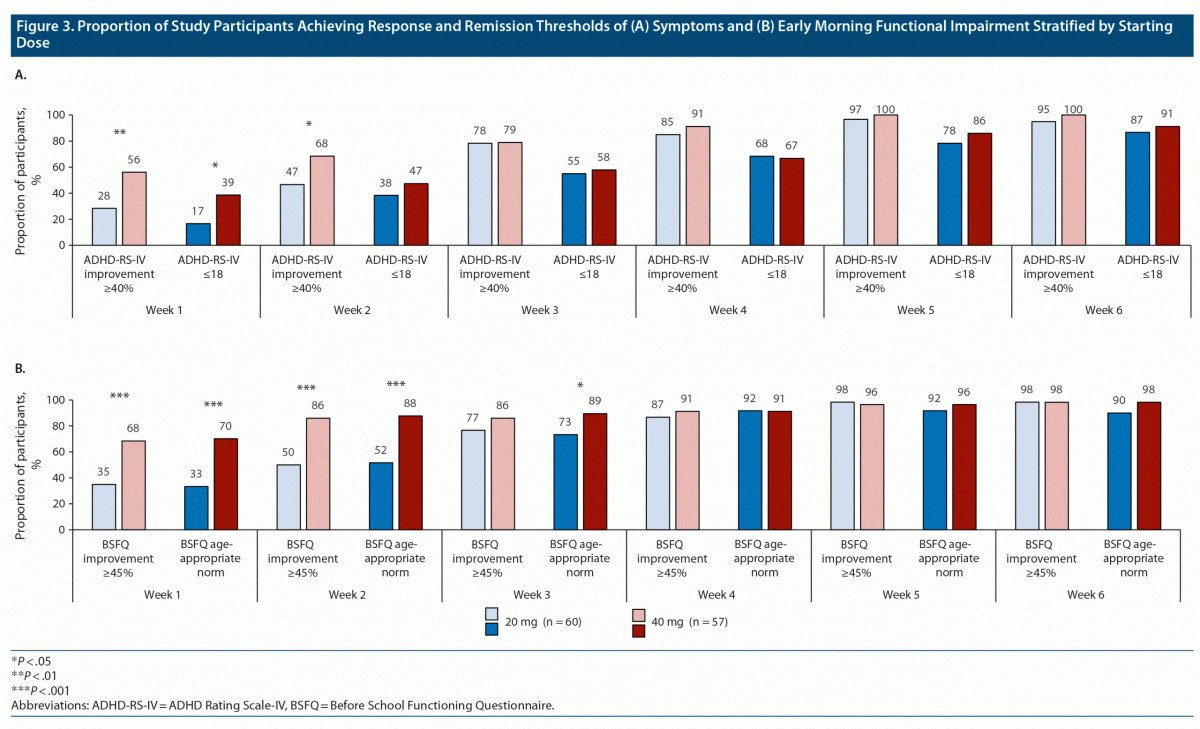
After 1 week of DR/ER-MPH treatment, participants with a starting dose of 40 mg versus 20 mg were more likely to achieve thresholds for symptomatic response by ADHD-RS-IV score (56% vs 28%; P = .0028), symptomatic remission by ADHD-RS-IV score (39% vs 17%; P = .0122), early morning functional response by BSFQ score (68% vs 35%; P = .0004), and early morning functional remission by BSFQ score (70% vs 33%; P < .0001) (Figure 3A and Figure 3B). After 2 weeks of DR/ER-MPH treatment, participants who started on 40 mg versus 20 mg were still more likely to achieve symptomatic response (68% vs 47%, P = .0246), early morning functional response (86% vs 50%; P < .0001), or early morning functional remission (88% vs 52%; P < .0001). By week 4 through to week 6, there were no longer significant differences in achievement of symptomatic or functional thresholds based on starting dose.
Adverse Events by Starting Dose
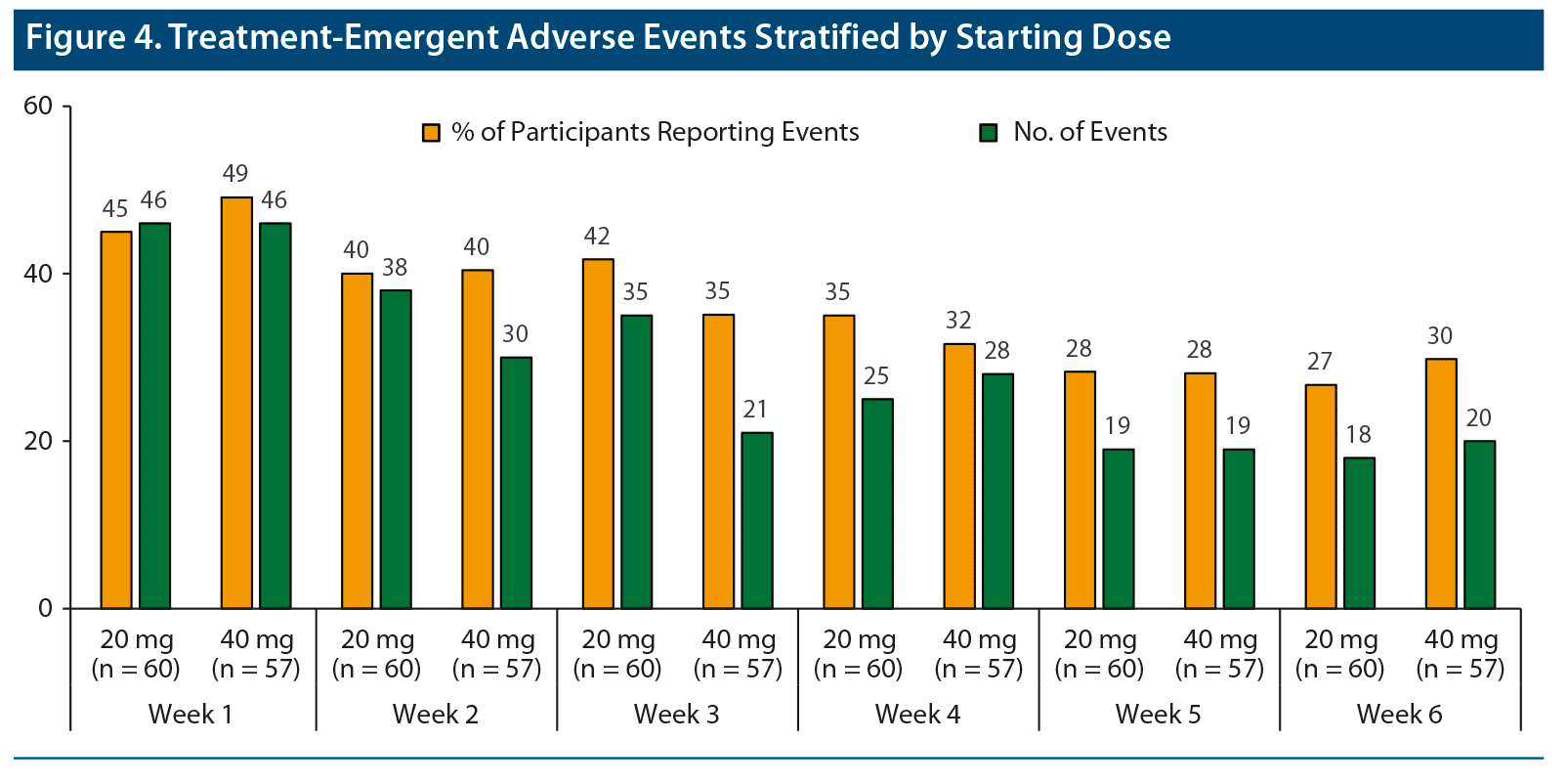
Safety results over the 6-week, open-label phase have been reported in detail elsewhere.12 As previously reported, the most common TEAEs (> 5%) during the open-label phase were any insomnia, decreased appetite, affect lability, headache, upper respiratory tract infection, upper abdominal pain, nausea or vomiting, increased diastolic blood pressure, tachycardia, and irritability.12 Three participants reported 5 TEAEs that led to the premature discontinuation of DR/ER-MPH during the open-label period: affect lability; aggression and agitation; and anxiety and panic attack.12 Among these 3 participants who discontinued, the starting dose was 20 mg in 2 participants and 40 mg in 1 participant. No serious TEAEs were reported during the open-label period or during the rest of the trial. When TEAEs were examined by starting dose, there were no obvious differences in the number or rate of TEAEs (Figure 4). For both starting doses, a larger proportion of participants reported TEAEs after the first week of treatment (45% and 49% for 20 and 40 mg, respectively) with a decreasing trend during the 6 weeks of treatment optimization (27% and 30%, respectively, during the sixth week of treatment). The types of TEAEs reported after 1 week of treatment were consistent with the overall TEAE profile; these early TEAEs did not prevent investigators from increasing doses for most participants (Table 1).
DISCUSSION
In this post hoc analysis of a phase 3 trial of DR/ER-MPH in children with ADHD, almost all participants at the end of the 6-week, open-label, DR/ER-MPH treatment optimization period achieved thresholds of clinically meaningful response and remission, respectively, on measures of symptoms based on the ADHD-RS-IV (97% and 89%), early morning functional impairment based on the BSFQ (98% and 94%) and PREMB-R AM (94% and 98%), and late afternoon/evening functional impairment based on the PREMB-R PM (91% and 84%) with a final optimized mean dose of 66.2 mg.
As described previously,20 the achievement of response (percentage improvement) and remission (endpoint score) thresholds must be considered in the context of baseline severity, as an individual with severe symptoms may achieve a threshold percentage improvement but still have impairing symptoms. On the other hand, an individual with mild symptoms or functional impairment may achieve remission but not achieve the percent improvement indicating response. These different scenarios are seen in this study: for the ADHD-RS-IV (Figure 1A) and PREMB-R PM (Figure 2B), the response thresholds were achieved at higher rates than remission thresholds; for the BSFQ, response and remission thresholds were achieved approximately equally (Figure 1B); for the PREMB-R AM, on which the mean baseline score indicated a severity of only screening risk, the remission threshold was achieved at a greater rate than response (Figure 2A). In contrast, mean BSFQ baseline scores indicated moderate severity in early morning functional impairment. The difference in baseline severities between the two scales is consistent with the previous hypothesis that the 20-item BSFQ is a more sensitive scale for determining severities of early morning functional impairment than the 3-item PREMB-R AM.30 Lower baseline ADHD-RS-IV and BSFQ scores were associated with numerically higher rates of response and remission over the first weeks of the dose-titration period; however, by week 6, similarly high rates of response and remission were achieved for participants with lower or higher than median baseline symptoms or early morning functional impairment (data not shown).
In the present study, rates of response and remission increased week-over-week (Figure 1) and roughly mirrored mean dose increases during the DR/ER-MPH optimization phase (Table 1), which corroborates previous studies that showed adequate dosing is necessary to achieve symptomatic remission.37,38 Previous studies have also shown that functional impairment does not necessarily improve at the same rate or to the same extent as symptoms.20,39 In the current study, higher rates of response and remission were achieved for early morning functional impairment compared to symptoms (Figure 1) during the first few weeks of the treatment-optimization period, indicating a robust morning effect even with low doses of DR/ER-MPH. However, even though 50% of participants achieved remission of early morning functional impairment after 1 week of treatment with their starting dose, the other participants required increased doses to achieve remission thresholds in the early morning, highlighting that dose titration can further optimize outcomes (Figure 1B). Because PREMB-R PM was not administered at every visit, it was not possible to evaluate how late afternoon/evening functional impairment decreased with DR/ER-MPH dose titration. Consistent with the dose-dependent duration of effect that has been predicted from pharmacokinetic/pharmacodynamic modeling,9 with higher doses increasing duration of effect mainly by extending evening efficacy, one might expect achievement of response and remission of late afternoon/evening functional impairment to require higher doses than for the early morning thresholds. Nevertheless, with optimized doses at week 6, remission rates were similarly high for symptoms (89%), early morning functional impairment (94%–98%), and late afternoon/evening functional impairment (84%), indicating control of ADHD symptoms and functional impairment throughout the day to the level of non-ADHD peers.
After 1 week of DR/ER-MPH treatment, mean ADHD-RS-IV and BSFQ scores improved beyond the 33% prespecified randomization criterion. Therefore, further dose adjustments were guided by weekly ADHD-RS-IV and BSFQ assessments as well as clinical judgment for optimizing control throughout the day. The final optimized DR/ER-MPH doses (mean = 66.2 mg) skewed toward the higher end of the approved dose range (20–100 mg) and were higher than what would have been predicted solely from bioavailability differences between formulations, due to the extended window of exposure from morning to the evening with DR/ER-MPH.36 Notably, investigators in this trial did not have access to the normative data or response thresholds for the BSFQ, PREMB-R AM, or PREMB-R PM. However, the recent identification of these thresholds adds tools that can be implemented to aid optimization of treatment for ADHD over the duration of the day. The norm-referenced severity cutoffs used to determine remission thresholds for early morning and late afternoon/evening functional impairment are unique in two ways: (1) they reference normative scores in the population and are adjusted for age; and (2) although only the threshold indicating children below screening risk was applied in this study, the multiple severity (percentile) cutoffs (screening risk [< 80th], mild [≥ 80th], moderate [≥ 93rd], severe [≥ 98th]) identified in the normative study30 allow clinicians to monitor incremental improvements, to optimize dosing (Supplementary Table 1), and to target further improvement. Furthermore, the results presented here using response and remission thresholds can provide them with a degree of predictability before initiating treatment.
Starting dose significantly affected the achievement of early response and remission on both measures of symptoms and measures of early morning functional impairment (Figure 3). In this study, investigators could prescribe a starting dose of either 20 mg or 40 mg of DR/ER-MPH based on previous treatment history, and approximately half of participants were started on each dose (Table 1). After 1 and 2 weeks of DR/ER-MPH treatment, significantly higher rates of symptomatic and early morning functional impairment thresholds were achieved in participants who started with a 40-mg dose versus a 20-mg dose (Figure 3), indicating a quicker and more robust response for the higher staring dose without an effect on TEAE rates (Figure 4). By the end of the 6-week treatment-optimization period, participants who started on 20 mg achieved similar symptomatic and functional outcomes compared to those who initially received 40 mg, suggesting that with careful monitoring and dose optimization almost complete response and remission were achieved regardless of starting dose. TEAEs were more prevalent early (Figure 4), but they rarely led to discontinuation or prevented dose increases (Table 1), indicating tolerability of treatment during dose optimization.
Selecting doses associated with improved rates of response and remission may be especially important considering that dose optimization in clinical practice may not occur at weekly intervals or be based on weekly administered rating scales, as was done in this study. Indeed, fewer than half of children prescribed medication have contact with their pediatrician within the first month of prescribing.40 Here, dose optimization based on weekly assessment of the entire day resulted in most children achieving response and remission thresholds for both symptoms and functional impairment at the bookends of the day by 6 weeks. The recent availability of these thresholds provides objective targets that may improve optimization of treatment for ADHD.
Interpretation of the study is limited by its post hoc nature and the treatment-optimization phase being open-label, which could have biased ratings but also reflects clinical experience. As mentioned previously,12 the inclusion of only methylphenidate responders with few comorbidities may limit generalization of findings. The analysis was limited to a 6-week treatment-optimization phase, and therefore long-term duration of response and remission remains to be tested prospectively.
In summary, the post hoc analyses reported here demonstrate that previously reported statistically significant mean improvements translate into clinically meaningful individual outcomes, with the majority of participants achieving response and remission of ADHD symptoms and early morning and late/afternoon functional impairment after 6 weeks when doses were appropriately titrated (final optimized dose was 66.2 mg). Improved early response and remission rates (by ADHD-RS-IV and BSFQ thresholds) were seen with a 40-mg versus a 20-mg starting dose without an increase in TEAEs. The results presented here are consistent with symptom and functional response and remission being realistic and achievable outcome goals with DR/ER-MPH when doses are optimized for control of ADHD symptoms and functional impairment throughout the day.
Clinical Points
- Results from clinical trials do not always generate the full complement of information that is relevant to clinicians, such as providing dosing recommendations to achieve optimal outcomes.
- After a 6-week dose optimization period with delayed-release and extended-release methylphenidate (DR/ER-MPH), clinically meaningful improvements were seen in attention-deficit/hyperactivity disorder (ADHD) symptom and functional impairment response and remission rates from early morning to evening in the vast majority of patients. The higher starting dose of 40 mg was associated with earlier improvements without sacrificing safety.
- Optimization of ADHD symptom control in the evening is especially important, as evening improvement lags behind morning improvement during titration due to the dose-dependent duration of effect of DR/ER-MPH. As doses were optimized overall outcomes improved, and optimized doses led to clinically meaningful evening improvements.
Submitted: February 1, 2021; accepted April 30, 2021.
Published online: June 22, 2021.
Author contributions: All authors had full access to all of the data in the study and had full responsibility for the content of the manuscript for publication. The corresponding author was responsible for the final review and had final responsibility for the decision to submit for publication.
Potential conflicts of interest: Dr Childress has received research support from Aevi Genomic Medicine; Akili Interactive Laboratories; Allergan; Arbor; Emalex; Forest; Ironshore Pharmaceuticals & Development, Inc.; KemPharm; Neos Therapeutics; Neurovance; Otsuka; Pearson; Pfizer; Purdue; Rhodes; Servier; Shire; Sunovion; Supernus; Takeda; Tris Pharma; and US Food and Drug Administration; serves on the advisory boards of Adlon; Akili Interactive Laboratories; Arbor; Cingulate Therapeutics; Ironshore Pharmaceuticals & Development, Inc.; Neos Therapeutics; Neurovance; NLS Pharma; Otsuka America; Pfizer; Purdue; Rhodes; Shire; Sunovion; Supernus; Takeda; and Tris Pharma; has consulted for Arbor; Ironshore Pharmaceuticals & Development, Inc.; Jazz; KemPharm; Neos Therapeutics; Neurovance; Purdue; Rhodes; Sunovion; Supernus Pharmaceuticals, Inc.; and Tris Pharma; has participated in speaker bureaus for Arbor; Ironshore Pharmaceuticals Inc.; Neos Therapeutics; Pfizer; Shire; Takeda; and Tris Pharma; and has received writing support from Arbor; Ironshore Pharmaceuticals & Development, Inc., Neos Therapeutics; Pfizer; Purdue; Rhodes; Shire; Sunovion; Takeda ; and Tris Pharma. Dr Cutler has received research support from Aevi Genomic Medicine; Akili Interactive Laboratories; Allergan; Arbor; Ironshore Pharmaceuticals & Development, Inc.; KemPharm; Lundbeck; Neos Therapeutics; Noven; Otsuka America; Purdue; Rhodes; Shire; Sunovion; Supernus; Takeda; and Tris Pharma; has served on the advisory board or as a consultant for Aevi Genomic Medicine; AiCure; Akili Interactive Laboratories; Allergan; Arbor; Atentiv; Cingulate; Ironshore Pharmaceuticals & Development, Inc.; KemPharm; Lundbeck; MedAvante-ProPhase; Neos Therapeutics; NLS Pharma; Noven; Otsuka America; Purdue; Rhodes; Shire; Sunovion; Supernus; Takeda; and Tris Pharma; has participated in speaker bureaus for AbbVie; Allergan; Arbor Pharmaceuticals; Ironshore Pharmaceuticals Inc.; Neos Therapeutics; Noven; Otsuka America; Shire; Sunovion; Supernus; Takeda; and Tris Pharma; and is a board member for the Neuroscience Education Institute. Dr Po is an employee of Highland Therapeutics Inc. Dr Warrington is an employee of Ironshore Pharmaceuticals Inc. Dr Sallee is an employee of Ironshore Pharmaceuticals Inc. and serves on the advisory board/board of directors of PD Bioscience. Mr DeSousa and Dr Incledon are employees of Ironshore Pharmaceuticals & Development, Inc.
Funding/support: This study was supported by Ironshore Pharmaceuticals & Development, Inc., Camana Bay, Grand Cayman, Cayman Islands.
Role of the sponsor: Ironshore Pharmaceuticals & Development, Inc., paid for the collection and analysis of data. Employees of Ironshore Pharmaceuticals & Development, Inc., and its affiliated companies were involved in the design of the study and interpretation of results and, therefore, are included as authors.
Previous presentations: This work has been presented in part at the 66th Annual Meeting of the American Academy of Child and Adolescent Psychiatry, Chicago, Illinois, October 14–19, 2019 • Academy of Managed Care (AMCP) Nexus Meeting, National Harbor, Maryland, October 29–November 1, 2019 • 25th Nevada Psychiatric Association Psychopharmacology Update, Las Vegas, Nevada, February 12–15, 2020 • The American Professional Society of ADHD and Related Disorders (APSARD), January 15–17, 2021 Virtual Conference.
Acknowledgements: The authors would like to acknowledge the statistical support from Hong Chen, MSc, at McDougall Scientific Ltd, supported by Ironshore Pharmaceuticals & Development, Inc., and the editorial assistance of Cassandra L. Uchida, PhD, and Marina Komolova, PhD, both of Highland Therapeutics Inc. and Justin Barnes, PhD, and Victor Otcheretko, MD, both of Ironshore Pharmaceuticals Inc.
Supplementary material: Available at Psychiatrist.com.
Editor’s Note: We encourage authors to submit papers for consideration as a part of our Focus on Childhood and Adolescent Mental Health section. Please contact Karen D. Wagner, MD, PhD, at [email protected].
References (40)

- Wolraich ML, Hagan JF Jr, Allan C, et al; Subcommittee on Children and Adolescents With Attention-Deficit/Hyperactive Disorder. Clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics. 2019;144(4):e20192528. PubMed CrossRef
- Canadian ADHD Practice Guidelines. 4.1 Edition. CADDRA website. https://www.caddra.ca/wp-content/uploads/CADDRA-ADHD-Practice-Guidelines-4.1-English.pdf. Updated 2020. Accessed October 22, 2020.
- Epstein JN, Weiss MD. Assessing treatment outcomes in attention-deficit/hyperactivity disorder: a narrative review. Prim Care Companion CNS Disord. 2012;14(6):PCC.11r01336. PubMed
- Brown TE, Flood E, Sarocco P, et al. Unmet medication coverage needs among adults with attention deficit/hyperactivity disorder (ADHD). Psychopharmacol Bull. 2017;47(4):18–28. PubMed
- Sallee FR. Early morning functioning in stimulant-treated children and adolescents with attention-deficit/hyperactivity disorder, and its impact on caregivers. J Child Adolesc Psychopharmacol. 2015;25(7):558–565. PubMed CrossRef
- Faraone SV, Schachar RJ, Barkley RA, et al. Early morning functional impairments in stimulant-treated children with attention-deficit/hyperactivity disorder versus controls: impact on the family. J Child Adolesc Psychopharmacol. 2017;27(8):715–722. PubMed CrossRef
- Jornay PM [package insert]. Camana Bay, Grand Cayman: Ironshore Pharmaceuticals & Development, Inc.; 2021.
- Vertzoni M, Augustijns P, Grimm M, et al. Impact of regional differences along the gastrointestinal tract of healthy adults on oral drug absorption: An UNGAP review. Eur J Pharm Sci. 2019;134:153–175. PubMed CrossRef
- Gomeni R, Komolova M, Incledon B, et al. Model-based approach for establishing the predicted clinical response of a delayed-release and extended-release methylphenidate for the treatment of attention-deficit/hyperactivity disorder. J Clin Psychopharmacol. 2020;40(4):350–358. PubMed CrossRef
- Childress A, Mehrotra S, Gobburu J, et al. Single-dose pharmacokinetics of HLD200, a delayed-release and extended-release methylphenidate formulation, in healthy adults and in adolescents and children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2018;28(1):10–18. PubMed CrossRef
- Pliszka SR, Wilens TE, Bostrom S, et al. Efficacy and safety of HLD200, delayed-release and extended-release methylphenidate, in children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2017;27(6):474–482. PubMed CrossRef
- Childress AC, Cutler AJ, Marraffino A, et al. A randomized, double-blind, placebo-controlled study of HLD200, a delayed-release and extended-release methylphenidate, in children with attention-deficit/hyperactivity disorder: an evaluation of safety and efficacy throughout the day and across settings. J Child Adolesc Psychopharmacol. 2020;30(1):2–14. PubMed CrossRef
- Swanson JM, Kraemer HC, Hinshaw SP, et al. Clinical relevance of the primary findings of the MTA: success rates based on severity of ADHD and ODD symptoms at the end of treatment. J Am Acad Child Adolesc Psychiatry. 2001;40(2):168–179. PubMed CrossRef
- Guideline on the clinical investigation of medicinal products for the treatment of attention deficit hyperactivity disorder (ADHD). European Medicines Agency website. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-attention-deficit-hyperactivity_en.pdf. Updated 2010. Accessed October 22, 2020.
- Findling RL, Childress AC, Krishnan S, et al. Long-term effectiveness and safety of lisdexamfetamine dimesylate in school-aged children with attention-deficit/hyperactivity disorder. CNS Spectr. 2008;13(7):614–620. PubMed CrossRef
- Cutler AJ, Brams M, Bukstein O, et al. Response/remission with guanfacine extended-release and psychostimulants in children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2014;53(10):1092–1101. PubMed CrossRef
- Kollins SH, Jain R, Brams M, et al. Clonidine extended-release tablets as add-on therapy to psychostimulants in children and adolescents with ADHD. Pediatrics. 2011;127(6):e1406–e1413. PubMed CrossRef
- Goodman D, Faraone SV, Adler LA, et al. Interpreting ADHD rating scale scores: linking ADHD rating scale scores and CGI levels in two randomized controlled trials of lisdexamfetamine dimesylate in ADHD. Prim Psychiatry. 2010;17(3):44–52.
- Dittmann RW, Cardo E, Nagy P, et al. Treatment response and remission in a double-blind, randomized, head-to-head study of lisdexamfetamine dimesylate and atomoxetine in children and adolescents with attention-deficit hyperactivity disorder. CNS Drugs. 2014;28(11):1059–1069. PubMed CrossRef
- Weiss M, Childress A, Mattingly G, et al. Relationship between symptomatic and functional improvement and remission in a treatment response to stimulant trial. J Child Adolesc Psychopharmacol. 2018;28(8):521–529. PubMed CrossRef
- Mattingly GW, Weisler RH, Young J, et al. Clinical response and symptomatic remission in short- and long-term trials of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. BMC Psychiatry. 2013;13:39. PubMed
- Newcorn JH, Sutton VK, Zhang S, et al. Characteristics of placebo responders in pediatric clinical trials of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2009;48(12):1165–1172. PubMed CrossRef
- Dickson RA, Maki E, Gibbins C, et al. Time courses of improvement and symptom remission in children treated with atomoxetine for attention-deficit/hyperactivity disorder: analysis of Canadian open-label studies. Child Adolesc Psychiatry Ment Health. 2011;5:14. PubMed
- Wigal TL, Newcorn JH, Handal N, et al. A double-blind, placebo-controlled, phase II study to determine the efficacy, safety, tolerability and pharmacokinetics of a controlled release (CR) formulation of mazindol in adults with DSM-5 attention-deficit/hyperactivity disorder (ADHD). CNS Drugs. 2018;32(3):289–301. PubMed CrossRef
- Weiss M, Childress A, Nordbrock E, et al. Characteristics of ADHD symptom response/remission in a clinical trial of methylphenidate extended release. J Clin Med. 2019;8(4):461. PubMed CrossRef
- Steele M, Jensen PS, Quinn DMP. Remission versus response as the goal of therapy in ADHD: a new standard for the field? Clin Ther. 2006;28(11):1892–1908. PubMed CrossRef
- Ramos-Quiroga JA, Casas M. Achieving remission as a routine goal of pharmacotherapy in attention-deficit hyperactivity disorder. CNS Drugs. 2011;25(1):17–36. PubMed CrossRef
- Mattingly G, Culpepper L, Babcock T, et al. Aiming for remission in adults with attention-deficit/hyperactivity disorder: the primary care goal. Postgrad Med. 2015;127(3):323–329. PubMed CrossRef
- Wilens TE, Faraone SV, Hammerness PG, et al. Clinically meaningful improvements in early morning and late afternoon/evening functional impairment in children with ADHD treated with delayed-release and extended-release methylphenidate. J Atten Disord. 2021 Jun 4;10870547211020073. PubMed. 10.1177/10870547211020073. Online ahead of print
- Faraone SV, DeSousa NJ, Komolova M, et al. Functional impairment in youth with ADHD: Normative data and norm-referenced cutoff points for the before school functioning questionnaire and the parent rating of evening and morning behavior scale, revised. J Clin Psychiatry. 2019;81(1):19m12956. PubMed CrossRef
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. Arlington, VA: American Psychiatric Association; 2013.
- Faraone SV, Hammerness PG, Wilens TE. Reliability and validity of the Before-School Functioning Scale in children with ADHD. J Atten Disord. 2018;22(11):1040–1048. PubMed CrossRef
- Faraone SV, Childress A, Wigal SB, et al. Reliability and validity of the Daily Parent Rating of Evening and Morning Behavior Scale, Revised. J Atten Disord. 2018;22(11):1066–1073. PubMed CrossRef
- Sutton V, Sumner C, Allen AJ, et al. Validity, reliability, and responsiveness of the DPREMB-R scale for ADHD. In: Abstracts of Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 2003; Miami Beach, FL.
- DuPaul GJ, Power TJ, Anastopoulos AD, et al. ADHD Rating Scale—IV: Checklists, norms, and clinical interpretation. New York, NY: Guilford Press; 1998.
- Childress AC, Uchida CL, Po MD, et al. A post hoc comparison of prior ADHD medication dose and optimized delayed-release and extended-release methylphenidate dose in a pivotal phase III trial. Clin Ther. 2020;42(12):2332–2340. PubMed CrossRef
- Greenhill LL, Swanson JM, Vitiello B, et al. Impairment and deportment responses to different methylphenidate doses in children with ADHD: the MTA titration trial. J Am Acad Child Adolesc Psychiatry. 2001;40(2):180–187. PubMed CrossRef
- Stein MA, Sarampote CS, Waldman ID, et al. A dose-response study of OROS methylphenidate in children with attention-deficit/hyperactivity disorder. Pediatrics. 2003;112(5):e404. PubMed CrossRef
- Coghill DR, Joseph A, Sikirica V, et al. Correlations between clinical trial outcomes based on symptoms, functional impairments, and quality of life in children and adolescents with ADHD. J Atten Disord. 2017;23(13):1578–1591. PubMed CrossRef
- Epstein JN, Kelleher KJ, Baum R, et al. Variability in ADHD care in community-based pediatrics. Pediatrics. 2014;134(6):1136–1143. PubMed CrossRef
This PDF is free for all visitors!




