Background: Individuals with Williams syndrome, a rare genetic disorder, are characterized by specific medical, cognitive, and behavioral phenotypes and often have high anxiety levels as well as phobia. Studies of the psychiatric phenotype in adults affected by Williams syndrome or literature on the management of their mental pathologies are lacking.
Method: In this article, we report the neuropsychiatric profile of 2 adult patients with Williams syndrome who also have generalized anxiety disorder and depressive symptoms (DSM-IV-TR criteria), along with their anxiety profiles and the strategies that were adopted for pharmacologic intervention.
Results: Neuropsychiatric profiles revealed a prefrontal cortex affliction that includes an alteration in executive functions. The patients had high scores for trait-anxiety and responded to treatment with a low-potency antipsychotic. A selective serotonin reuptake inhibitor (SSRI) was coadministered with the antipsychotic to alleviate the depressive symptoms. The treatment led to an improvement in self-control, mental concentration, and social skills, as well as decreased irritability and aggressiveness and stabilization of mood.
Conclusions: The combination of SSRIs and low doses of low-potency antipsychotics seems to be the most suitable medication to treat generalized anxiety disorder and related disorders in individuals with Williams syndrome. Manic reactions and increase in anxiety must be closely monitored during treatment. Control of anxiety and sleep should be a priority in these patients, even as a preventative measure.
Prim Care Companion CNS Disord 2013;15(4):doi:10.4088/PCC13m01504
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: January 17, 2013; accepted March 26, 2013.
Published online: August 1, 2013.
Corresponding author: Tania Ramos-Moreno, PhD, Department of Clinical Sciences, Division of Neurology, Experimental Epilepsy Group, Wallenberg Neuroscience Center, Lund University, Lund, Sweden ([email protected]).
Neuropsychiatric and Behavioral Profiles of 2 Adults With Williams Syndrome: Response to Antidepressant Intake
ABSTRACT
Background: Individuals with Williams syndrome, a rare genetic disorder, are characterized by specific medical, cognitive, and behavioral phenotypes and often have high anxiety levels as well as phobia. Studies of the psychiatric phenotype in adults affected by Williams syndrome or literature on the management of their mental pathologies
are lacking.
Method: In this article, we report the neuropsychiatric profile of 2 adult patients with Williams syndrome who also have generalized anxiety disorder and depressive symptoms (DSM-IV-TR criteria), along with their anxiety profiles and the strategies that were adopted for pharmacologic intervention.
Results: Neuropsychiatric profiles revealed a prefrontal cortex affliction that includes an alteration in executive functions. The patients had high scores for trait-anxiety and responded to treatment with a low-potency antipsychotic. A selective serotonin reuptake inhibitor (SSRI) was coadministered with the antipsychotic to alleviate the depressive symptoms. The treatment led to an improvement in self-control, mental concentration, and social skills, as well as decreased irritability and aggressiveness
and stabilization of mood.
Conclusions: The combination of SSRIs and low doses of low-potency antipsychotics seems to be the most suitable medication to treat generalized anxiety disorder and related disorders in individuals with Williams syndrome. Manic reactions and increase in anxiety must be closely monitored during treatment. Control of anxiety and sleep
should be a priority in these patients, even
as a preventative measure.
Prim Care Companion CNS Disord 2013;15(4):doi:10.4088/PCC13m01504
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: January 17, 2013; accepted March 26, 2013.
Published online: August 1, 2013.
Corresponding author: Tania Ramos-Moreno, PhD, Department of Clinical Sciences, Division of Neurology, Experimental Epilepsy Group, Wallenberg Neuroscience Center, Lund University, Lund, Sweden
([email protected]).
Williams syndrome is a rare genetic disorder affecting 1 in 25,000 to 50,000 births, although new prevalence estimates are as high as 1 in 7,500.1 Williams syndrome is caused by a hemizygous deletion of ~ 1.6 megabases, containing ~ 28 genes, on chromosome 7q11.23.2,3 The classical presentation of Williams syndrome has referred to the well-known clinical features (growth retardation, cardiovascular abnormalities, and infantile hypercalcemia),4–6 and neurologic problems have recently been added to the clinical features (coordination difficulties, hyperreflexia, hyperacusis).7–9
This syndrome is characterized by a mild-to-moderate mental retardation and a relative preservation of linguistic and social-affective skills alongside relative strengths in auditory rote memory and facial recognition. These features contrast with the profound impairment of visual-spatial abilities and numeric processing.7–12 Since this syndrome is characterized by striking verbal and musical features and by impairment of visual-spatial abilities, which have been subject to many studies,11–13 the psychiatric symptoms7,14–19 have been largely ignored by researchers and medical doctors. Individuals with Williams syndrome have also been strongly associated with high anxiety levels, as well as phobia development,17,18 hyperacusis,12 attention-deficit/hyperactivity disorder (ADHD; 70% of individuals with Williams syndrome have been diagnosed with ADHD),7,14,15 and related psychological symptoms such as poor concentration.16
Recently, more information about psychiatric medication intake by patients affected by Williams syndrome has been demanded,20 and new evidence suggests that an alteration in executive functions such as working memory and attention shifting exists in these individuals.21–23 In this article, we report the neuropsychiatric profiles of 2 adult patients with Williams syndrome who also have generalized anxiety disorder, and we characterize their anxiety profiles and the strategies that were adopted for pharmacologic intervention. We found that the patients were responsive to selective serotonin reuptake inhibitors (SSRIs), suggesting that the combination of SSRIs and low doses of antipsychotics is the most suitable medication to be given in these cases. The patients presented to Clinica San Juan de Dios, Madrid, Spain. Patients and caregivers provided consent, first, to allow treatment and, second, to publish the findings.
CASE REPORTS
Mr A
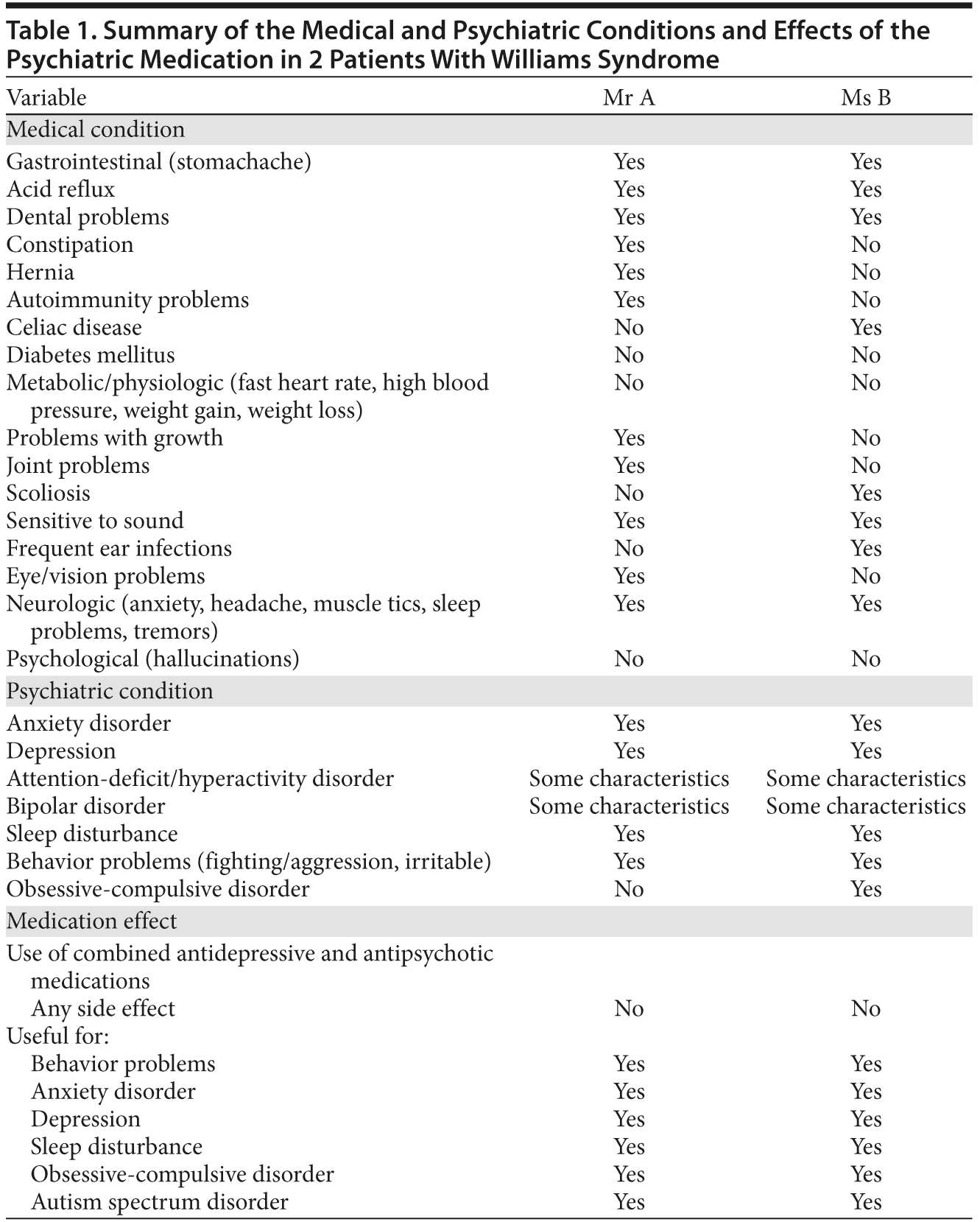
Mr A, a 26-year-old man, was diagnosed with Williams syndrome at the age of 11 years, with confirmed positive fluorescence in situ hybridization to an elastin gene deletion in chromosome 7. Mr A lives with his mother, is dependent for activities of daily living, and attends an occupational center on weekday mornings. Mr A is unlikely to integrate in the workforce, except in jobs specially designed for him. He needs supervision to take care of household chores, although he can perform most of them. Mr A also has difficulties in making and maintaining friendships and needs to be supervised by his family when trying to be autonomous in social relationships. The quality of his leisure time is low and unsatisfactory, with no hobbies apart from going to shopping malls to try to have social contact. In this regard, he has difficulties in respecting social norms, exhibiting behaviors such as intensive eye contact that becomes obsessive and problematic and especially with regard to excessive proximity to strangers. He also is quite irritable in the family environment and presents a mild decline in sexual conduct, with an intense dissatisfaction. In addition, Mr A suffers from various disorders and diseases that can be framed within the Williams syndrome diagnosis, such as chronic pain conditions and gastrointestinal complaints, including chronic gastritis and gastroesophageal reflux (Table 1).
Mr A fulfills DSM-IV-TR criteria for generalized anxiety disorder. The onset of this disorder can be dated from the age of 13 years until present. Although Mr A has a slight worsening of anxiety symptoms during spring and autumn, he fails to meet the criteria for seasonal affective disorder. Mr A also fulfilled DSM-IV-TR criteria for a mild depressive state when he first came to the clinic. This state was characterized by a decrease in his ability to concentrate, increased self-absorption, apathy and lack of motivation, stereotyped movements, and intensified obsessive symptoms, such as repetitive speech and a marked intensity in his compulsive behavior.
Independently from Williams syndrome, Mr A has also been diagnosed with Behcet’s syndrome, an autoimmune disease characterized by recurrent mucocutaneous lesions and sight threatening uveitis that may involve the central nervous system.24 His family estimates the first appearance of symptoms at the age of 13 years. Interestingly, they also report that he may have experienced a mild depressive state when he was 12 to 13 years old.
Ms B
Ms B, a 35-year-old woman, was diagnosed with Williams syndrome at the age of 20 years, with confirmed positive fluorescence in situ hybridization to an elastin gene deletion in chromosome 7. She lives with her mother and is dependent for activities of daily living; she is unable to work outside the home or do household chores unsupervised, although she can perform most of them. Ms B is unable to maintain friendships outside the family even though she unwinds well in a known social environment, with apparently no major difficulties in respecting social norms apart from effusively approaching strangers and excessive visual contact, which becomes problematic. Ms B has hobbies, including playing the piano and attending piano classes. She also shows a mild decline in sexual conduct.
Ms B’s medical disorders framed within the Williams syndrome organic symptoms include dyspepsia and postprandial acute pain associated with gastroesophageal reflux. Ms B has developed a phobia to certain types of foods, as she suffered from celiac disease and was not diagnosed for years. The phobia to foods has resolved. The celiac disease is currently being treated, and, thus, Ms B eats gluten-free foods.
With regard to psychiatric disorders, Ms B has a specific phobia to swallow, fulfilling DSM-IV-TR criteria for phobias, leading to weight loss. Moreover, she presented with increased anxiety episodes that fulfill DSM-IV-TR criteria for generalized anxiety disorder and irritability that fulfills DSM-IV-TR criteria for a mixed state with mild depressive symptoms and dysphoria, characterized by anhedonia, reduced motivation and initiative, neglect of normal activities (piano lessons, outings with family and friends), and insomnia with increased sleep-onset latency and early wake-up. In addition, Ms B currently fulfills DSM-IV-TR criteria for a mild obsessive-compulsive disorder (OCD) (Table 1).
NEUROPSYCHIATRIC
PROFILES AND ANXIETY
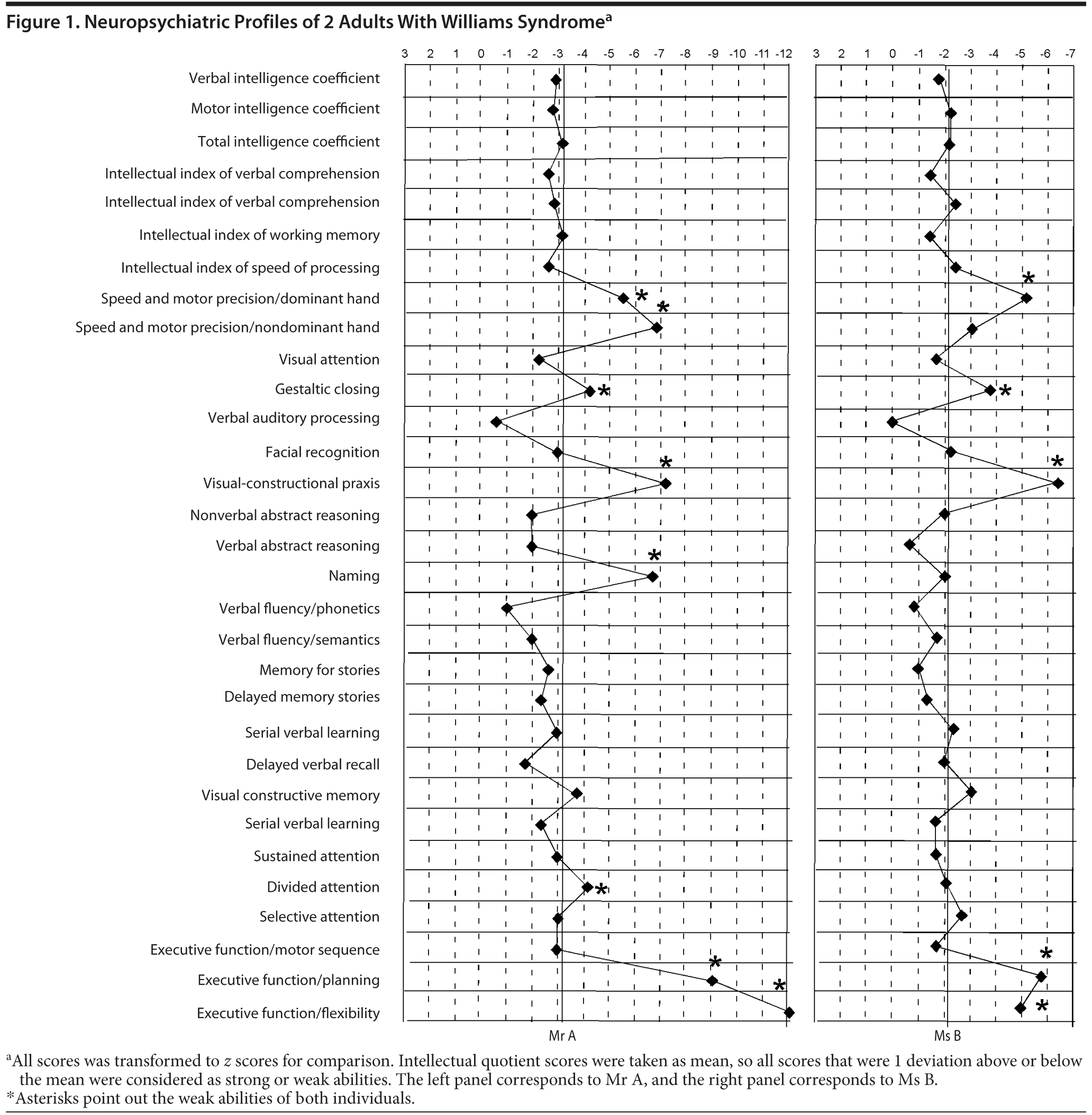
Both patients were given the following tests: the Weschler Adult Intelligence Scale, Third Edition (WAIS-III)25; Test of Memory and Learning26; Benton Facial Recognition Test27; Trail Making Test parts A and B28; verbal fluency test29; Purdue Pegboard Test30; Rey-Osterrieth Complex Figure Test31; Woodcock-Μuñoz Battery32; and the Stroop task.33 All scores were transformed to z scores for comparison. The total intellectual quotient (IQ) score was calculated as a mean, so if the score for a given test was 1 or more deviations above the total IQ score, the feature was considered as a strong ability and vice versa. Mr A’s total IQ score per the WAIS-III was 56. Ms B’s total IQ score per the WAIS-III was 66.
Auditory verbal processing, abstract verbal reasoning, phonetic verbal fluency, and memory for stories scored as peaks for both patients and fit within the strong abilities described for individuals with Williams syndrome.14,34 Gestaltic closing, visual-constructional praxis, and executive functions were the valleys of their abilities, as have been previously suggested.22,23,35 All other processes were adjusted to the level expected given the patients’ overall intellectual level (Figure 1).
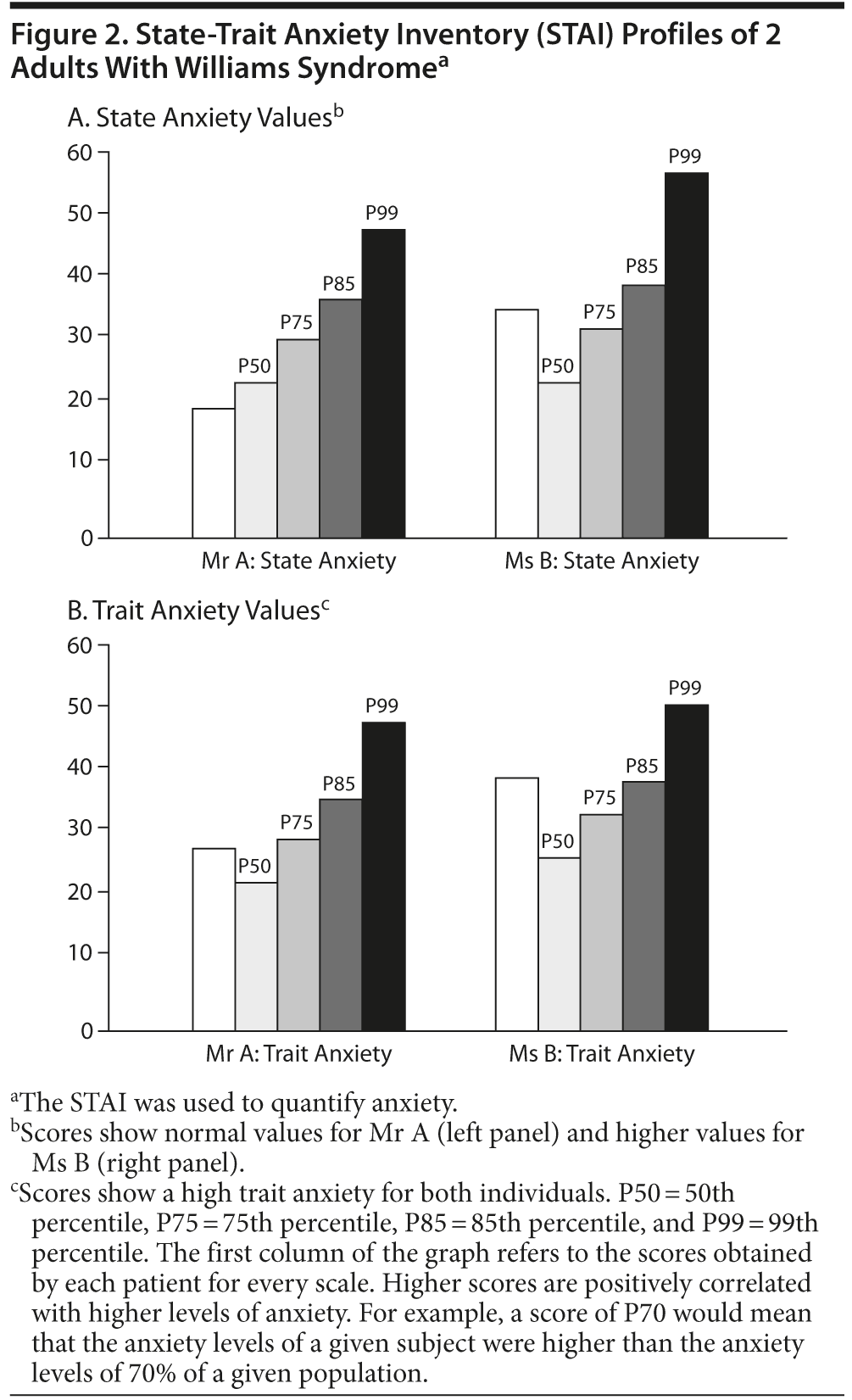
In addition to their neuropsychiatric profile, we also assessed the patients’ emotional state with the State-Trait Anxiety Inventory (STAI)36 and Beck Depression Inventory (BDI).37 Both patients were in a depressive state when they first started treatment. Mr A’s BDI results indicate that he was not currently in a depressive episode, although his score could be interpreted as at risk for depression. Ms B’s BDI results are suggestive of depressive state I. The STAI profile reveals that both patients have a high score for anxiety as a feature. This finding was expected on the basis of previous literature.17 Mr A improved his anxiety state to reach normal values, probably due to his medical treatment. Ms B’s anxiety symptoms slowly decreased to baseline values (Figure 2).
Psychotropic Response
Mr A
For the last 3 years, Mr A has been maintained with a combination of low-potency sedative antipsychotics (levomepromazine oral drops 21 mg/d) and very low doses of an SSRI (citalopram 10 mg/d), well below the effective doses used for adults, which tend to be in the range of 20–60 mg/d. The variations of his treatment are summarized in Table 2.
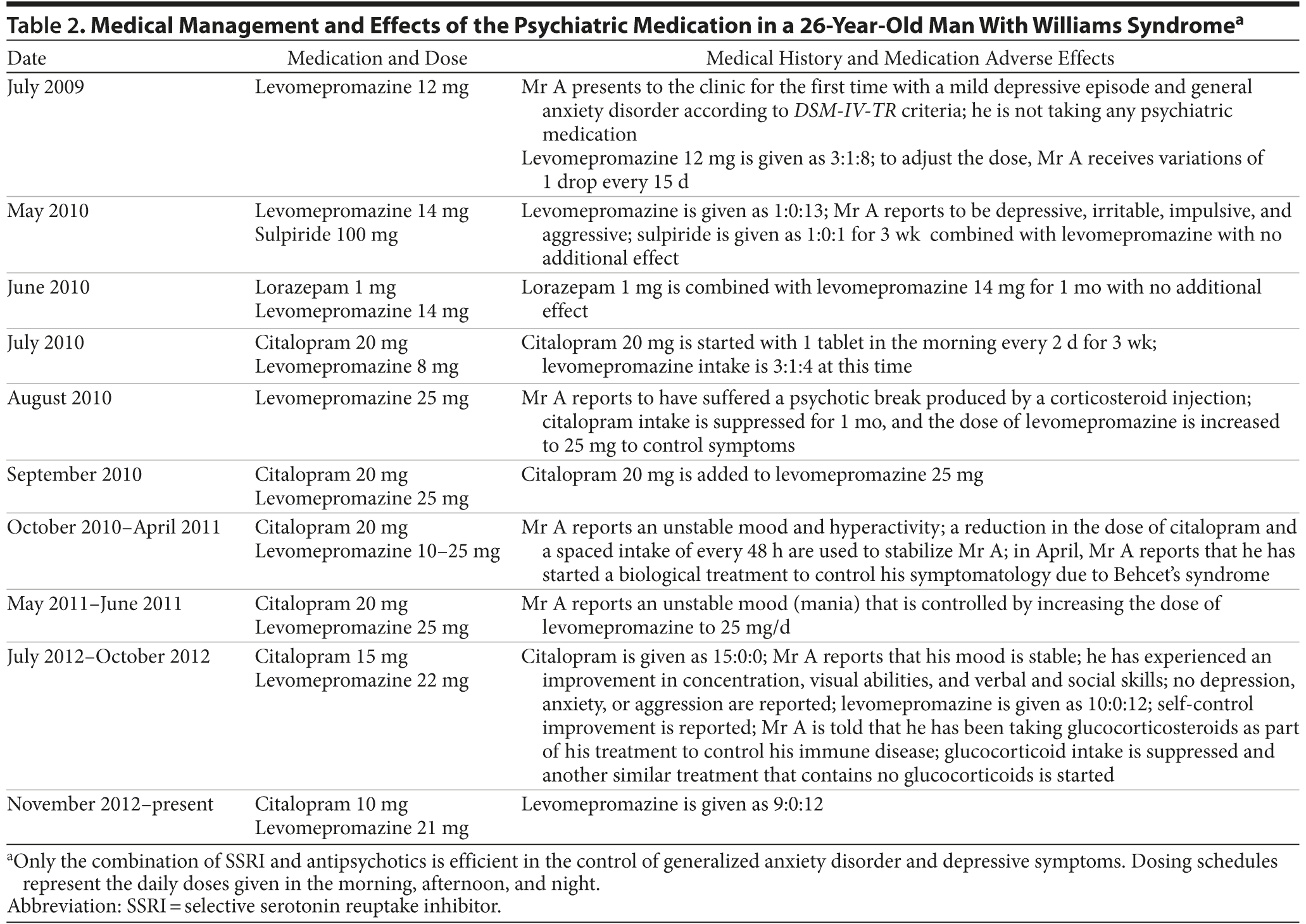
Treatment with levomepromazine was initiated, aiming for an improvement in Mr A’s anxiety and hypomanic state (irritability, aggressiveness). Once these symptoms were under control, a mild depressive state arose and was treated with the use of citalopram, as trials with the benzodiazepine lorazepam and sulpiride did not yield any improvements in the depressive state. The mild depressive state disappeared within 2 weeks following the SSRI intake (citalopram 20 mg/d). After this treatment, Mr A experienced a short hypermanic state, encompassing an increase of uninhibited sexual behavior and irritability that lasted less than 1 week. The citalopram was reduced to the current maintenance dose of 10 mg/d along with levomepromazine 21 mg/d. Low doses of levomepromazine are maintained to counterbalance the side effects (hypomania) of SSRI intake and to help his anxiety and sleeping problems. It is noteworthy to say that Mr A has remarkably improved in autonomy and social skills since the beginning of treatment.
Mr A did not experience adverse effects in response to the SSRI treatment, apart from 1 occasion when he suffered a psychotic episode, most likely in response to an intramuscular bolus dose of corticosteroids (methylprednisolone 40 mg) administered to control a mild anaphylactic shock of unknown etiology. This episode was regulated by suppressing the SSRI for 1 month and increasing the dose of levomepromazine to 25 mg. We did not aim at that time to improve the psychotic symptoms (paranoid and megalomaniac poorly structured delusions, increased disorganization of speech), as these symptoms were assumed to be an acute response to the corticosteroids.
In addition, since Mr A has the autoimmune disease Behcet’s syndrome, unknown to the psychiatrist, he was treated with corticosteroids for months, which resulted in mood variations (such as irritability, depressed mood, anxiety) as expected from a hypercortisolemic state.38,39 Interestingly, corticosteroids have been suggested to act directly on the serotoninergic system.40 In order to control these mood variations, the dose range of levomepromazine varied between 10 and 25 mg/d (maximum). Although the antipsychotic doses of levomepromazine given to normal adults are usually higher (over 100 mg/d), we adjusted our dose range based on the observation of clinical outcomes both to regulate the sleep and to alleviate the irritability and dysphoria. Mr A currently reports that his mood has been stabilized. Depression, irritability, and aggressiveness are under control. He has also experienced an improvement in self-control, concentration, and visual, verbal, and social skills.
Ms B
Ms B is currently being treated with a combination of sedative antipsychotics (levomepromazine oral drops
25 mg/24 h) and low doses of an SSRI (citalopram 20 mg/24 h), which may be low to produce drastic changes but are enough for maintenance. The variations of her treatment are summarized in Table 3.
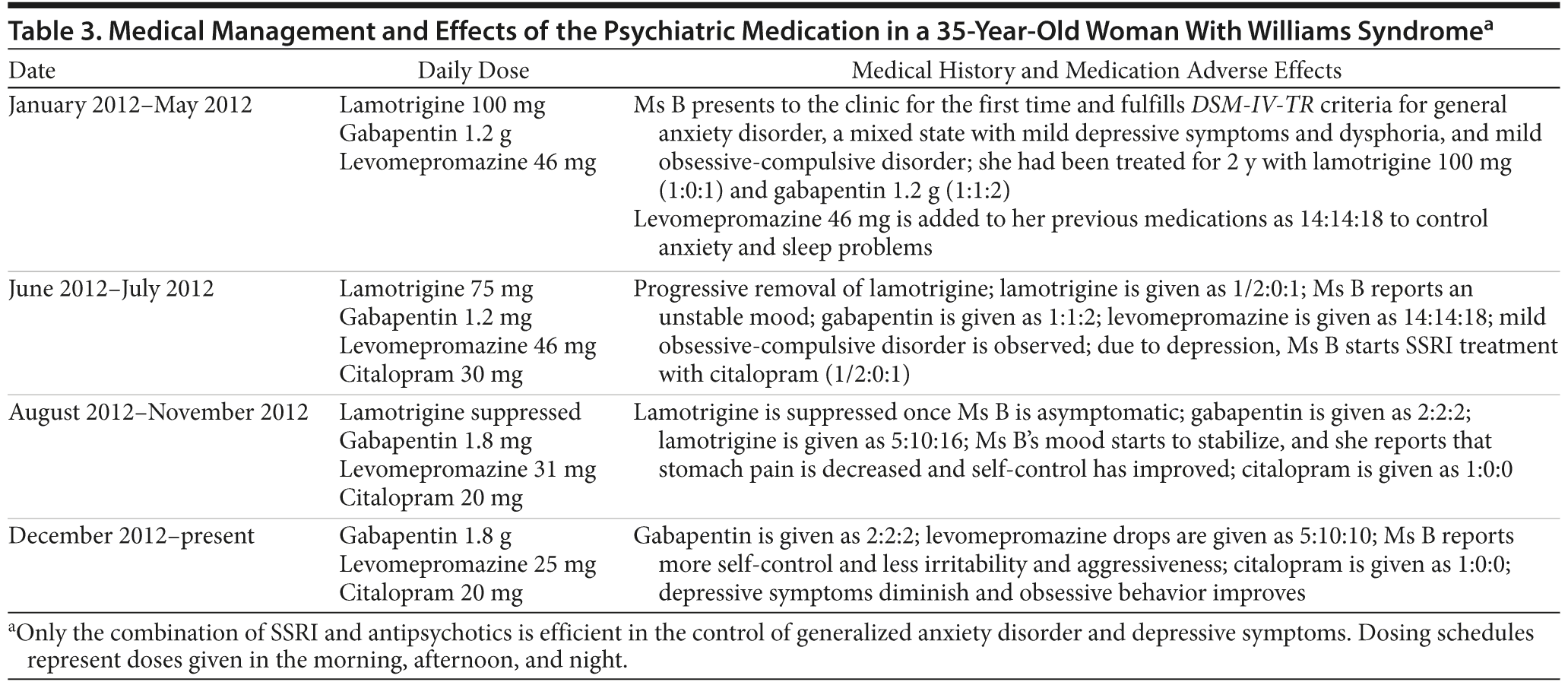
When Ms B first came to the clinic, she was being treated for depression with lamotrigine every 12 hours and gabapentin. Treatment with levomepromazine was initiated, aiming to improve her anxiety and sleep problems. The depressive state appeared only after the anxiety symptoms were gone, and, thus, we included citalopram 30 mg in her treatment. Once depressive symptoms subsided, lamotrigine treatment was stopped. Gabapentin has not yet been retired, as it maintains the strength of its effect with time and helps with anxiety (Ms B takes 1.8 g/d, 600 mg every 6 h). Ms B currently reports that she has experienced an improvement in self-control, irritability, and aggressiveness, and her mood has been stabilized. She feels happier than before, although she still has problems controlling her obsessions.
We conclude that the SSRI therapy seems to be the most adequate treatment to control mood variations and generalized anxiety disorder in individuals with Williams syndrome. However, treatment should be administered in lower doses than what is typically prescribed. The medication dose must be fine-tuned to each patient and supplemented with low doses of a typical, low-potency antipsychotic such as levomepromazine.
DISCUSSION
Williams Syndrome: Psychiatric Profile
and Response to Antidepressants
The most common psychiatric disorders found in Williams syndrome have been reported mainly in children and encompass ADHD in 65%–84%,10,41 specific phobia in 43%–54%,41,42 and generalized anxiety disorders in 8%–24% of the cases.41,42 Longitudinal trajectories of anxiety disorders in children with Williams syndrome have been evaluated, and it has been suggested that anxiety suffered by these children is most often chronic.43
Despite the difference in age and gender, both individuals with Williams syndrome presented here match in terms of strengths and weaknesses, and the neuropsychiatric profiles reveal a prefrontal cortex affliction in both subjects. A loss of prefrontal cortex abilities can give rise to an impulse control disinhibition. Interestingly, impulse control disorder has been described in individuals with Williams syndrome17 (T.R.M., personal observation). Impulse control disorder is considered to be part of the OCD spectrum and has also been described as a trait in bipolar disorder. Thus, a common biological basis may be behind both OCD and bipolar disorder pathologies.44,45 Patients with OCD tend to have an imbalance of serotonin and dopamine levels in the prefrontal cortex.46 Drugs that increase serotonin output, such as SSRIs, reduce symptoms of OCD and are effective in bipolar disorder.47–49 We postulate that it is likely that Williams syndrome neurologic pathologies stem from a similar biological basis as bipolar disorder, which is further validated by the finding that patients with Williams syndrome are highly responsive to SSRIs, as are patients with bipolar disorder.49 In addition, an imbalance in serotonin receptors has been reported in the prefrontal cortex of a Williams syndrome mouse model50 and has been linked to the pathogeny of bipolar disorder.51
Features involving the prefrontal cortex such as executive functions (planning and mental flexibility) have so far been poorly studied in Williams syndrome.22,23 Here, we confirm and extend previous data on the executive functions as well as the decreased visual abilities. Furthermore, we have studied features such as delayed verbal recall; serial visual learning; sustained, divided, and selective attention and motor sequencing; intellectual index of processing speed; intellectual index of perceptive organization; and denomination and delayed memory for stories and found them to be in accordance with the intellectual level of the patients.
Williams Syndrome: Digestive System,
Autoimmunity and Anxiety
Digestive problems including gastroesophageal reflux are well-known and encompassed in Williams syndrome.19 Gastrointestinal problems might cause a deficiency in the immune system and could result in autoimmune problems in the long-term, as happens in other diseases such as Parkinson’s disease.52,53 Moreover, an inflammation in the digestive system could be potentiating a dysregulation in the hypothalamic pituitary system52 that could in turn radicalize anxiety and OCD in the long-term. A dysregulation in the hypothalamic pituitary axis has recently been described in Williams syndrome,54 and to our knowledge, no studies describing the existence of autoimmune diseases in patients suffering from Williams syndrome are available. The above data, together with the higher frequency of celiac disease in patients with Williams syndrome19 and previous data showing alterations in abnormal B-cell development,55 suggest that a positive feedback might be occurring in Williams syndrome, ultimately enhancing the symptoms of anxiety. Untreated or mistreated anxiety could ultimately develop psychopathologic aspects in Williams syndrome in the long-term, and these neglected aspects have been described in those with Williams syndrome.19
CONCLUSION
More detailed information with regard to treating and characterizing anxiety in Williams syndrome is needed for psychiatrists and neuropsychologists. Consideration of anxiety and frontal lobe affectations as basic symptoms in patients affected by Williams syndrome could help professionals and caregivers in the detection of these symptoms and would prevent treatments that potentiate psychopathologies in the long-term. Moreover, treating anxiety properly in Williams syndrome might allow these patients to develop adequate responses to social stimuli and neuropsychological strategies. The combination of SSRIs and low doses of antipsychotics to counteract SSRI side effects seems to be the most suitable medication to treat generalized anxiety disorder and related disorders in individuals with Williams syndrome.
Drug names: citalopram (Celexa and others), gabapentin (Neurontin and others), lamotrigine (Lamictal and others), lorazepam (Ativan and others), methylprednisolone (Depo-Medrol, A-Methapred, and others).
Author affiliations: Clinica San Juan de Dios and Clinica Nuestra Señora de La Paz, Madrid, Spain (Dr Urgeles and Ms Alonso); Department of Molecular Biology and Center of Molecular Biology “Severo Ochoa,” Universidad Autónoma de Madrid, Cantoblanco, Madrid, Spain; and Department of Clinical Sciences, Division of Neurology, Experimental Epilepsy Group, Wallenberg Neuroscience Center, Lund University,
Lund, Sweden (Dr Ramos-Moreno).
Potential conflicts of interest: Dr Ramos-Moreno has received grant/research support from the Spanish Ministry of Economy and Competitiveness (formerly Science and Innovation, PLE2009-0101, SAF2010-17167). Dr Urgeles and Ms Alonso report no financial or other affiliations relevant to the subject of this article.
Funding/support: None reported.
Acknowledgments: The authors thank Concepcion Fournier, PhD, Hospital Infantil Niño Jesús, Madrid, Spain, for her help with the neuropsychiatric profile study; Agnieszka Krzyzanowska, PhD, Hospital 12 de Octubre, Madrid, Spain, for the revision of the manuscript; and Alberto Martinez-Serrano, PhD, Center of Molecular Biology “Severo Ochoa,” Madrid, Spain, for his fruitful discussions. Drs Fournier, Krzyzanowska, and Martinez-Serrano report no conflicts of interest related to the subject of this article.
REFERENCES
1. Strømme P, Bjørnstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17(4):269–271. doi:10.1177/088307380201700406 PubMed
2. Baumer A, Dutly F, Balmer D, et al. High level of unequal meiotic crossovers at the origin of the 22q11. 2 and 7q11.23 deletions. Hum Mol Genet. 1998;7(5):887–894. doi:10.1093/hmg/7.5.887 PubMed
3. Valero MC, de Luis O, Cruces J, et al. Fine-scale comparative mapping of the human 7q11.23 region and the orthologous region on mouse chromosome 5G: the low-copy repeats that flank the Williams-Beuren syndrome deletion arose at breakpoint sites of an evolutionary inversion(s). Genomics. 2000;69(1):1–13. doi:10.1006/geno.2000.6312 PubMed
4. Williams JC, Barratt-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation. 1961;24(6):1311–1318. doi:10.1161/01.CIR.24.6.1311 PubMed
5. Beuren AJ, Apitz J, Harmjanz D. Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation. 1962;26(6):1235–1240. doi:10.1161/01.CIR.26.6.1235 PubMed
6. Morris CA. In Morris CA, Lenhoff, HM, and Wang PP, eds. Williams–Beuren Syndrome: Research, Evaluation, and Treatment. Baltimore, MD: Johns Hopkins University Press; 2006:3–17
7. Pober BR, Dykens EM. Williams syndrome: an overview of medical, cognitive, and behavioural features. Child Adolesc Psychiatr Clin N Am. 1996;5:929–943.
8. Mervis CB, Robinson BF, Pani JR. Visuospatial construction. Am J Hum Genet. 1999;65(5):1222–1229. doi:10.1086/302633 PubMed
9. Mervis CB, Klein-Tasman BP. Williams syndrome: cognition, personality,
and adaptive behavior. Ment Retard Dev Disabil Res Rev. 2000;6(2):148–158. doi:10.1002/1098-2779(2000)6:2<148::AID-MRDD10>3.0.CO;2-T PubMed
10. Morris CA, Demsey SA, Leonard CO, et al. Natural history of Williams syndrome: physical characteristics. J Pediatr. 1988;113(2):318–326. doi:10.1016/S0022-3476(88)80272-5 PubMed
11. Mervis CB. In Morris CA, Lenhoff HM, Wang PP, eds. Williams–Beuren Syndrome: Research, Evaluation, and Treatment. Baltimore, MD: Johns Hopkins University Press, 2006
12. Meyer-Lindenberg A, Mervis CB, Berman KF. Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition
and behaviour. Nat Rev Neurosci. 2006;7(5):380–393. doi:10.1038/nrn1906 PubMed
13. Laws G, Bishop D. Pragmatic language impairment and social deficits in Williams syndrome: a comparison with Down’s syndrome and specific language impairment. Int J Lang Commun Disord. 2004;39(1):45–64. doi:10.1080/13682820310001615797 PubMed
14. Bellugi U, Bihrle A, Jernigan T, et al. Neuropsychological, neurological,
and neuroanatomical profile of Williams syndrome. Am J Med Genet suppl. 1990;6:115–125. PubMed
15. Dilts CV, Morris CA, Leonard CO. Hypothesis for development of a behavioral phenotype in Williams syndrome. Am J Med Genet suppl. 1990;6:126–131. PubMed
16. Lashkari A, Smith AK, Graham JM Jr. Williams-Beuren syndrome:
an update and review for the primary physician. Clin Pediatr (Phila). 1999;38(4):189–208. doi:10.1177/000992289903800401 PubMed
17. Dykens EM. Anxiety, fears, and phobias in persons with Williams syndrome. Dev Neuropsychol. 2003;23(1–2):291–316. doi:10.1080/87565641.2003.9651896 PubMed
18. Blomberg S, Rosander M, Andersson G. Fears, hyperacusis and musicality
in Williams syndrome. Res Dev Disabil. 2006;27(6):668–680. doi:10.1016/j.ridd.2005.09.002 PubMed
19. Morris CHaWP. Williams Beuren Syndrome. Research, Evaluation and Treatment. Baltimore, MD: The John Hopkins University Press; 2006.
20. Martens MA, Seyfer DL, Andridge RR, et al. Parent report of antidepressant, anxiolytic, and antipsychotic medication use in individuals with Williams syndrome: effectiveness and adverse effects. Res Dev Disabil. 2012;33(6):
2106–2121. doi:10.1016/j.ridd.2012.06.006 PubMed
21. Rhodes SM, Riby DM, Park J, et al. Executive neuropsychological functioning in individuals with Williams syndrome. Neuropsychologia. 2010;48(5):
1216–1226. doi:10.1016/j.neuropsychologia.2009.12.021 PubMed
22. Osório A, Cruz R, Sampaio A, et al. How executive functions are related to intelligence in Williams syndrome. Res Dev Disabil. 2012;33(4):1169–1175. doi:10.1016/j.ridd.2012.02.003 PubMed
23. Menghini D, Addona F, Costanzo F, et al. Executive functions in individuals with Williams syndrome. J Intellect Disabil Res. 2010;54(5):418–432. doi:10.1111/j.1365-2788.2010.01287.x PubMed
24. Pleyer U, Hazirolan D, Winterhalter S, et al. Behçet’s disease—ophthalmological and general aspects, part 1: etiology, pathogenesis and diagnostics. Ophthalmologe. 2012;109(11):1129–1141, quiz 1142–1143. doi:10.1007/s00347-012-2698-5 PubMed
25. Wechsler D. Wechsler Adult Intelligence Scale. San Antonio, TX: Psychological Corporation; 1997.
26. Reynolds CR, Voress JK. Test of Memory and Learning. Austin, TX:
Pro-Ed; 2007.
27. Benton A. The Revised Visual Retention Test. New York, NY: Psychological Corporation; 1974.
28. Corrigan JDHM, Hinkeldey NS. Relationships between parts A and B
of the Trail Making Test. J Clin Psychol. 1987;43(4):402–409. doi:10.1002/1097-4679(198707)43:4<402::AID-JCLP2270430411>3.0.CO;2-E PubMed
29. Benton ALHK. Multilingual Aphasia Examination Manual. Iowa City, IA: University of Iowa; 1976.
30. Tiffin J, Asher EJ. The Purdue pegboard; norms and studies of reliability
and validity. J Appl Psychol. 1948;32(3):234–247. doi:10.1037/h0061266 PubMed
31. Rey A. L’examen clinique en psychologique. Arch Psychol. 1941;28:112–164.
32. Muñoz-Sandoval AF, Woodcock RW, McGrew KS, et al. Batería III Woodcock-Muñoz. Itasca, IL: Riverside Publishing; 2005.
33. Stroop JR. Studies of interference in serial verbal reactions. J Exp Psychol. 1935;18(6):643–662. doi:10.1037/h0054651
34. Trauner DA, Bellugi U, Chase C. Neurologic features of Williams and
Down syndromes. Pediatr Neurol. 1989;5(3):166–168. doi:10.1016/0887-8994(89)90066-0 PubMed
35. Farran EK. Perceptual grouping ability in Williams syndrome: evidence for deviant patterns of performance. Neuropsychologia. 2005;43(5):815–822. doi:10.1016/j.neuropsychologia.2004.09.001 PubMed
36. Spielberger CD, Gorssuch RL, Lushene PR, et al. Manual for the State-Trait Anxiety Inventory. Palo Alto, CA: Consulting Psychologists Press, Inc; 1983.
37. Beck AT. Depression: Causes and Treatment. Philadelphia: University of Pennsylvania Press; 2006.
38. Starkman MN, Schteingart DE, Schork MA. Depressed mood and other psychiatric manifestations of Cushing’s syndrome: relationship to hormone levels. Psychosom Med. 1981;43(1):3–18. PubMed
39. Wolkowitz OM, Burke H, Epel ES, et al. Glucocorticoids: mood, memory,
and mechanisms. Ann N Y Acad Sci. 2009;1179(1):19–40. doi:10.1111/j.1749-6632.2009.04980.x PubMed
40. McAllister-Williams RH, Ferrier IN, Young AH. Mood and neuropsychological function in depression: the role of corticosteroids
and serotonin. Psychol Med. 1998;28(3):573–584. doi:10.1017/S0033291798006680 PubMed
41. Leyfer OT, Woodruff-Borden J, Klein-Tasman BP, et al. Prevalence of psychiatric disorders in 4 to 16-year-olds with Williams syndrome.
Am J Med Genet B Neuropsychiatr Genet. 2006;141B(6):615–622. doi:10.1002/ajmg.b.30344 PubMed
42. Kennedy JC, Kaye DL, Sadler LS. Psychiatric diagnoses in patients with Williams syndrome and their families. Jefferson J Psychiatry. 2006;20(1):22–31.
43. Woodruff-Borden J, Kistler DJ, Henderson DR, et al. Longitudinal course
of anxiety in children and adolescents with Williams syndrome.
Am J Med Genet C Semin Med Genet. 2010;154C(2):277–290. doi:10.1002/ajmg.c.30259 PubMed
44. McElroy SL, Pope HG Jr, Keck PE Jr, et al. Are impulse-control disorders related to bipolar disorder? Compr Psychiatry. 1996;37(4):229–240. doi:10.1016/S0010-440X(96)90001-2 PubMed
45. Swann AC, Dougherty DM, Pazzaglia PJ, et al. Impulsivity: a link between bipolar disorder and substance abuse. Bipolar Disord. 2004;6(3):204–212. doi:10.1111/j.1399-5618.2004.00110.x PubMed
46. Clarke HF, Dalley JW, Crofts HS, et al. Cognitive inflexibility after prefrontal serotonin depletion. Science. 2004;304(5672):878–880. doi:10.1126/science.1094987 PubMed
47. Thorén P, Asberg M, Cronholm B, et al. Clomipramine treatment of obsessive-compulsive disorder, 1: a controlled clinical trial. Arch Gen Psychiatry. 1980;37(11):1281–1285. doi:10.1001/archpsyc.1980.01780240079009 PubMed
48. Maina G, Albert U, Salvi V, et al. Weight gain during long-term treatment of obsessive-compulsive disorder: a prospective comparison between serotonin reuptake inhibitors. J Clin Psychiatry. 2004;65(10):1365–1371. doi:10.4088/JCP.v65n1011 PubMed
49. Parker G, Tully L, Olley A, et al. SSRIs as mood stabilizers for bipolar II disorder? a proof of concept study. J Affect Disord. 2006;92(2–3):205–214. doi:10.1016/j.jad.2006.01.024 PubMed
50. Proulx E, Young EJ, Osborne LR, et al. Enhanced prefrontal serotonin 5-HT(1A) currents in a mouse model of Williams-Beuren syndrome
with low innate anxiety. J Neurodev Disord. 2010;2(2):99–108. doi:10.1007/s11689-010-9044-5 PubMed
51. Kishi T, Okochi T, Tsunoka T, et al. Serotonin 1A receptor gene, schizophrenia and bipolar disorder: an association study and meta-analysis. Psychiatry Res. 2011;185(1–2):20–26. doi:10.1016/j.psychres.2010.06.003 PubMed
52. Meisel C, Schwab JM, Prass K, et al. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6(10):775–786. doi:10.1038/nrn1765 PubMed
53. Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease.
Parkinsonism Relat Disord. 2011;17(1):10–15. doi:10.1016/j.parkreldis.2010.08.003 PubMed
54. Dai L, Carter CS, Ying J, et al. Oxytocin and vasopressin are dysregulated in Williams syndrome, a genetic disorder affecting social behavior. PLoS ONE. 2012;7(6):e38513. doi:10.1371/journal.pone.0038513 PubMed
55. Ranheim EA, Kwan HC, Reya T, et al. Frizzled 9 knock-out mice have abnormal b-cell development. Blood. 2005;105(6):2487–2494. doi:10.1182/blood-2004-06-2334 PubMed
Enjoy this premium PDF as part of your membership benefits!


