Objective: To examine the role of butyrylcholinesterase (BuChE) in cholinergic signaling and neurologic conditions, such as Alzheimer’s disease (AD). The rationale for inhibiting cholinesterases in the management of AD, including clinical evidence supporting use of the dual acetylcholinesterase (AChE) and BuChE inhibitor rivastigmine, is discussed.
Data Sources: PubMed searches were performed using butyrylcholinesterase as a keyword. English-language articles referenced in PubMed as of September 2011 were included.
Study Selection and Data Synthesis: English-language articles related to BuChE considered to be of clinical relevance to physicians were included. English-language articles specifically related to AChE were not included, as the role of AChE in cholinergic signaling and the underlying pathology of AD is well documented. Reference lists of included publications were used to supplement the search.
Results: AChE and BuChE play a role in cholinergic signaling; BuChE can hydrolyze acetylcholine and compensate for AChE when levels are depleted. In the AD brain, AChE levels decrease, while BuChE levels are reportedly increased or unchanged, with changes becoming more pronounced during the disease course. Furthermore, BuChE genotype may influence AD risk and rate of disease progression. Strategies that increase acetylcholine levels (eg, cholinesterase inhibitors) demonstrate symptomatic efficacy in AD. Rivastigmine has proven cognitive efficacy in clinical trials, and data suggest that its action is mediated, in part, by inhibition of BuChE. Retrospective analyses of clinical trials provide evidence that BuChE genotype may also influence treatment response.
Conclusions: AChE-selective inhibitors and a dual AChE and BuChE inhibitor demonstrate symptomatic efficacy in AD. Mounting preclinical and clinical evidence for a role of BuChE in maintaining normal cholinergic function and the pathology of AD provides a rationale for further studies investigating use of rivastigmine in AD and the influence of BuChE genotype on observed efficacy.
Prim Care Companion CNS Disord 2013;15(2):doi:10.4088/PCC.12r01412
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: May 16, 2012; accepted October 11, 2012.
Published online: March 7, 2013.
Corresponding author: Agneta Nordberg, MD, PhD, Karolinska Institutet, Alzheimer Neurobiology Center, Novum, 141 86 Stockholm, Sweden ([email protected]).
A Review of Butyrylcholinesterase as a Therapeutic Target
in the Treatment of Alzheimer’s Disease
ABSTRACT
Objective: To examine the role of butyrylcholinesterase (BuChE) in cholinergic signaling and neurologic conditions, such as Alzheimer’s disease (AD). The rationale for inhibiting cholinesterases in the management of AD, including clinical evidence supporting use of the dual acetylcholinesterase (AChE) and BuChE inhibitor rivastigmine, is discussed.
Data Sources: PubMed searches were performed using butyrylcholinesterase as a keyword. English-language articles referenced in PubMed as of September 2011 were included.
Study Selection and Data Synthesis: English-language articles related to BuChE considered to be of clinical relevance to physicians were included. English-language articles specifically related to AChE were not included, as the role of AChE in cholinergic signaling and the underlying pathology of AD is well documented. Reference lists of included publications were used to supplement the search.
Results: AChE and BuChE play a role in cholinergic signaling; BuChE can hydrolyze acetylcholine and compensate for AChE when levels are depleted. In the AD brain, AChE levels decrease, while BuChE levels are reportedly increased or unchanged, with changes becoming more pronounced during the disease course. Furthermore, BuChE genotype may influence AD risk and rate of disease progression. Strategies that increase acetylcholine levels (eg, cholinesterase inhibitors) demonstrate symptomatic efficacy in AD. Rivastigmine has proven cognitive efficacy in clinical trials, and data suggest that its action is mediated, in part, by inhibition of BuChE. Retrospective analyses of clinical trials provide evidence that BuChE genotype may also influence treatment response.
Conclusions: AChE-selective inhibitors and a dual AChE and BuChE inhibitor demonstrate symptomatic efficacy in AD. Mounting preclinical and clinical evidence for a role of BuChE in maintaining normal cholinergic function and the pathology of AD provides a rationale for further studies investigating use of rivastigmine in AD and the influence of BuChE genotype on observed efficacy.
Prim Care Companion CNS Disord 2013;15(2):doi:10.4088/PCC.12r01412
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: May 16, 2012; accepted October 11, 2012.
Published online: March 7, 2013.
Corresponding author: Agneta Nordberg, MD, PhD, Karolinska Institutet, Alzheimer Neurobiology Center, Novum, 141 86 Stockholm, Sweden ([email protected]).
Cholinergic neurotransmitter systems are widely distributed in the human brain and play an important role in regulating many processes, including memory, learning, attention, and behavior.1–3 Normal cholinergic function depends on the rapid hydrolysis of the neurotransmitter acetylcholine (ACh) by cholinesterases.4 The human brain contains 2 major cholinesterases: acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE).5 AChE is reported to be the more abundant of the 2 cholinesterases in the brain.5 However, there is increasing evidence that BuChE has an important role in cholinergic mediation.
In this review, we will consider the role of BuChE in normal cholinergic function and in pathological disease states, such as Alzheimer’s disease (AD). The rationale for inhibiting cholinesterases in the symptomatic treatment of AD and mounting clinical evidence supporting the use of a dual inhibitor of both AChE and BuChE are discussed.
METHOD
Relevant articles were identified by performing searches of PubMed with butyrylcholinesterase as a search term. Searches were performed in September 2011. English-language articles related to BuChE considered to be of clinical relevance to physicians were included. Articles specifically related to AChE were not included, as the role of AChE in cholinergic signaling and the underlying pathology of AD is well documented. References within those articles included were used to supplement the search.
RESULTS
Mechanism of Action of AChE and BuChE in the
Cholinergic Synapse and the Extrasynaptic Role
of ACh-Hydrolyzing Enzymes
In the normal human brain, nerve impulses in the presynaptic neuron result in fusion of ACh-containing vesicles with the presynaptic membrane and the release of ACh into the synaptic cleft.6 ACh diffuses across the synaptic cleft and interacts with cholinergic receptors on the postsynaptic neuron (Figure 1). AChE activity is important for terminating nerve impulses within the central nervous system (CNS) and maintaining pulsatile cholinergic stimulation. AChE is a serine hydrolase, which mediates the hydrolysis of ACh to choline and acetate (Figure 1).6 Choline is then transported back to the presynaptic neuron and is used as a substrate for synthesis of ACh.6 It is hypothesized that, in a manner analogous to the inactivation of glutamate in glutamatergic transmission, synaptically released ACh can also be hydrolyzed to choline and acetate by glial BuChE. Choline is then returned to the synaptic cleft for reuptake into cholinergic neurons (Figure 1).5,7
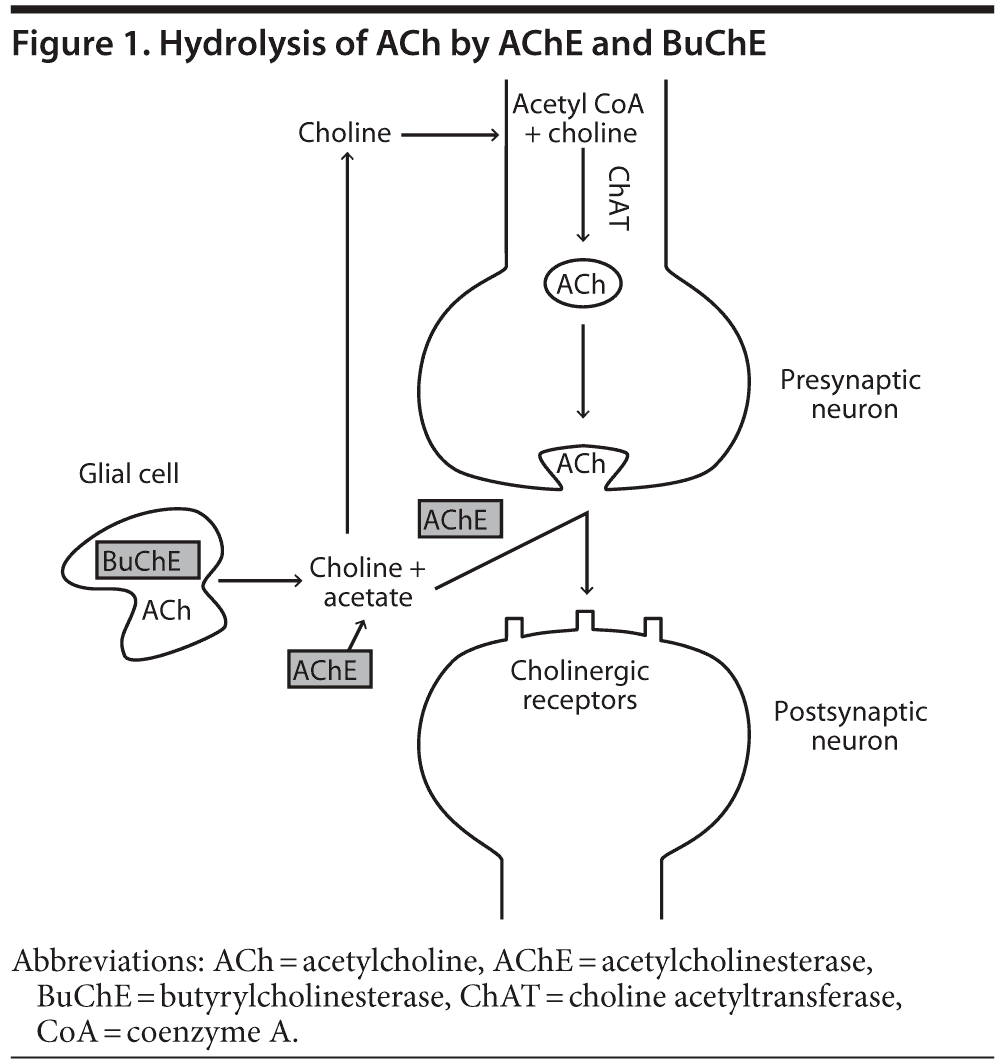
In addition to ACh activity at neuronal synaptic clefts, ACh activity is also detected in the extracellular fluid8 and cerebrospinal fluid (CSF),9 acting on nonexcitable cholinoceptive cells such as microglia, astrocytes, oligodendrocytes, and endothelial cells.10 BuChE may be the primary extrasynaptic ACh-hydrolyzing enzyme.11
Distribution of AChE and BuChE in the CNS
Immunohistochemical and in situ hybridization studies have investigated the distribution of choline acetyltransferase, a specific marker of cholinergic neurons, in the peripheral nervous system and CNS.12 Using these methods, cholinergic neurons have been detected in the striatum, basal forebrain, cerebral cortex, mesopontine tegmental nuclei, cranial motor nuclei, and spinal motor neurons.12
AChE is consistently associated with both cholinergic and cholinoceptive neurons. BuChE immunoreactivity has been detected in all brain regions using an enzyme-linked immunosorbent assay.13 AChE is expressed at particularly high levels in the hippocampus formation5,14 and the motor, premotor, and neocortical areas of the human cerebral cortex.15 BuChE is also expressed in the hippocampus and temporal neocortex, but at lower levels than AChE.5 Mesulam et al5 reported that hippocampal and neocortical AChE is localized in the axons and pyramidal neurons, while BuChE is associated with glial cells. However, Darvesh et al16 reported that, in the hippocampal formation, AChE is present in both neurons and neuropil, while BuChE is only detected in neurons, and suggested that these enzymes may colocalize. In the amygdala, the number of BuChE-positive neurons is reported to exceed the number of AChE-positive neurons, with BuChE residing predominantly in the neurons and their dendritic processes and AChE residing in the neuropil.16 The distinct distribution of AChE and BuChE within the brain suggests that these enzymes may both play important biological roles.
Elucidating the Roles of AChE
and BuChE in Cholinergic Signaling
Studies in AChE-knockout mice have investigated the role of AChE and BuChE in cholinergic signaling. Mice nullizygous for AChE show no AChE activity and normal BuChE activity but still appear to have structurally intact cholinergic pathways.17–19 Mesulam et al19 reported that both AChE-knockout and wild-type mice show BuChE activity in all parts of the brain that receive cholinergic innervations and demonstrated that BuChE could hydrolyze the acetylcholine surrogate acetylthiocholine. Hartmann et al20 reported a dose-dependent increase in ACh levels in wild-type mice, but not in AChE-knockout mice, treated with an AChE-selective inhibitor. In contrast, infusion of a BuChE-selective inhibitor resulted in elevation of ACh levels in AChE-knockout mice but had no effect on wild-type mice.20 Evidence for a role of BuChE in cholinergic signaling in humans comes from a study that demonstrated that BuChE can hydrolyze acetylthiocholine in human brain tissue treated with the AChE inhibitor BW-284C51.5 Together, these studies in AChE-knockout mice and human brain tissue have shown that BuChE can hydrolyze ACh and can compensate for AChE when levels are depleted.
In addition to having a role in the hydrolysis of ACh, BuChE is also known to have nonenzymatic functions. It has been suggested that AChE may accelerate amyloid deposition in the Alzheimer’s brain21 and that BuChE can associate with amyloid-β (Aβ) protein and may delay the onset and rate of neurotoxic Aβ fibril formation in vitro. 22 AChE and BuChE may also be involved in inflammatory pathways.23
Changes in AChE and BuChE Activity in Alzheimer’s Disease, Dementia With Lewy Bodies, and Parkinson’s Disease Dementia: The Cholinergic Hypothesis
AD is characterized by marked cholinergic dysfunction.24 More specifically, degeneration of cholinergic neurons; loss of cholinergic transmission; depletion of ACh, especially in moderate-to-severe disease stages; and changes in AChE and BuChE activity are commonly observed in the cerebral cortex and hippocampus of patients with AD.24,25 These factors are thought to contribute to disease progression,24,26 with degeneration of cholinergic circuits associated with the progressive impairment in memory and cognitive function that is typically observed.
AChE is thought to be the predominant cholinesterase in healthy human brains. However, decreased AChE activity and increased or unchanged BuChE activity have been observed in certain brain regions in AD.24,27 In patients with AD, BuChE activity is reported to be significantly higher in men than in women.28 In the AD brain, BuChE activity is associated with amyloid plaques and neurofibrillary tangles,29–32 with BuChE covering up to 6 times more of the plaque area compared with age-matched controls.33
Patients with Parkinson’s disease and Parkinson’s disease dementia show reduced AChE activity in the frontal cortex compared with healthy controls.34 Higher levels of AChE and BuChE are observed in patients with Parkinson’s disease dementia compared with Parkinson’s disease.34 In the CSF, AChE and BuChE levels were significantly higher in patients with Parkinson’s disease dementia compared with those with Parkinson’s disease but were similar between patients with Parkinson’s disease and controls.35 A further study reported that the CSF ratio of AChE:BuChE was lower in patients with Parkinson’s disease dementia compared with those with Parkinson’s disease, but AChE and BuChE activity was comparable.36 BuChE activity in the CSF has been shown to vary according to the severity or duration of dementia associated with Parkinson’s disease or the disability stage of the disease.36 No differences in BuChE activity were detected in the CSF and serum in demented patients with dementia with Lewy bodies, nondemented patients with dementia with Lewy bodies, and controls.37
Changes in Expression of AChE and BuChE
During the Course of Alzheimer’s Disease
In addition to changes in activity, changes in AChE and BuChE protein expression are reported to occur throughout the disease course. Both AChE and BuChE are known to exist in 6 polymeric forms divided into 2 classes, asymmetric and globular, based on the presence and absence of a collagen-like tail.38 The globular forms of AChE and BuChE may be further subdivided into G1, G2, and G4 forms.39 The G4 form is the most predominant form expressed in the CNS and is responsible for degradation of ACh at the cholinergic synapse.38 The G1 form is found in smaller amounts in the brain.38 As AD progresses, there is an increase in the G1 form of both AChE and BuChE and a decrease in the G4 form of AChE, particularly in the hippocampus and amygdala.39,40 Ogane et al41 reported a decrease in the membrane-bound G4 form of AChE by 71%, 45%, and 47% in the frontal cortex, parietal cortex, and caudate putamen, respectively, and a decrease in the G1, G2, and G4 aqueous soluble forms of AChE in the caudate putamen (unchanged in the frontal and parietal cortex) in patients with AD compared with controls. Increased levels of glial-derived BuChE and decreases in synaptic AChE result in a relative increase in the ratio of BuChE to AChE in cortical regions from approximately 0.6 to 11.42
High levels of BuChE in gray matter have been correlated with annual cognitive decline in a prospective, autopsy-confirmed population with dementia with Lewy bodies.43 However, high CSF BuChE activity may be associated with greater levels of cognitive function in patients with AD.28 It has been hypothesized that low levels of BuChE in the CSF may correlate with high levels of BuChE/AChE/Aβ/apolipoprotein E (APOE) complexes (BAβAC) in the vicinity of, or trapped within, plaques, along with cerebral amyloid angiopathy, increased neurotoxicity, and greater central neurodegeneration.10
The observed changes in BuChE activity and expression throughout the course of AD, and the relationship between BuChE levels and cognitive function, emphasize the potential value of BuChE in addition to AChE as a therapeutic target in patients with AD.
Influence of BuChE Genotype on Enzymatic Activity and the Rate of Disease Progression
BuChE-K is the most common polymorphism of BuChE and is found in up to one-third of Asians and Caucasians.44 The BuChE-K genotype arises from a DNA point mutation45 and is associated with approximately 30% lower BuChE activity than wild-type alleles.45 BuChE-K shows reduced ability to inhibit Aβ fibril formation.46 A higher load of cholinesterase-positive neuritic plaques has been reported in the cerebral cortex of BuChE-K carriers with late-onset AD.47 Postmortem analysis of 30 patients with autopsy-diagnosed dementia (AD or dementia with Lewy bodies) reported significantly less phosphorylated tau protein but no significant differences in Aβ protein in patients with ≥ 1 BuChE-K allele compared with wild-type alleles.48
Reports on the impact of BuChE genotype on the risk of developing AD are conflicting. It has been reported that BuChE-K is overrepresented among patients with AD and that carriers of BuChE-K show an increased risk of developing AD.49–52 A further study demonstrated that BuChE-K is associated with an increased risk of AD, but only in BuChE-K homozygotes ≥ 70 years of age.53 However, it has also been reported that BuChE-K is expressed at a lower frequency in patients with AD compared with healthy controls.54 Several studies have reported no significant difference in the frequency of BuChE-K alleles between patients with and without AD.55–61 A higher allelic frequency of BuChE-K has also been reported in patients with dementia with Lewy bodies relative to Parkinson’s disease dementia.62
Despite several studies reporting an association between BuChE-K and the risk of developing AD, it has been suggested that BuChE-K may actually protect against disease progression. A community-based study of 339 patients with severe AD found that the presence of a BuChE-K allele was associated with a slower average rate of cognitive decline (Mini-Mental State Examination [MMSE] score).63 Differing profiles in cognitive test performance have also been reported in a study comparing healthy volunteers with silent or wild-type BuChE alleles, providing further evidence of a role for BuChE in cognitive function.64
Influence of BuChE and APOE Epsilon 4 Genotype
on Risk of Developing Alzheimer’s Disease
APOE epsilon 4 (APOE ε4) is a commonly reported genetic risk factor for AD.65 High levels of APOE protein have been associated with low cerebral glucose metabolism, high cerebral Aβ load, and phosphorylated tau in the CSF, while BuChE levels were found to have the opposite relationship.10 A further study reported an association between high levels of APOE protein and low levels of BuChE.66 CSF BuChE activity is reported to be 40% to 60% higher in APOE ε4–negative patients than in those with 1 or 2 APOE ε4 alleles.28
Data suggest a complex interplay between BuChE and APOE ε4 genotype and the risk of developing AD. In carriers of APOE ε4, wild-type BuChE has been reported to be associated with an increased risk of developing AD.67 Hiltunen et al68 reported a lower frequency of BuChE-K in AD patients under the age of 75 years who were also carriers of APOE ε4 compared with nondemented controls. Other studies have reported an association between BuChE-K and APOE ε4 and the risk of developing late-onset AD.50,51,69,70 Sodeyama et al55 reported a significant association of BuChE-K with the neuropathological changes of AD in carriers of APOE ε4, but not in noncarriers, over the age of 75 years. Another study showed that, in the presence of APOE ε4, carriers of BuChE-K showed a dose-dependent reduction in CSF BuChE activity compared with noncarriers.71 However, in the absence of APOE ε4, BuChE activity was essentially indistinguishable between carriers of BuChE-K and noncarriers.71 A further study reported that, in non–APOE ε4 carriers, BuChE-K may protect against AD but only in women.72
The rate of AD progression may be influenced by both APOE ε4 and BuChE genotype. A post hoc exploratory analysis of a 3- to 4-year, randomized, placebo-controlled trial of rivastigmine in patients with mild cognitive impairment showed that the rate of progression to AD and loss of hippocampal volume were highest in carriers of both APOE ε4 and BuChE-K and lowest in carriers of BuChE-K without APOE ε4.73 In support of these findings, Darreh-Shori et al71 observed that in a study of patients with AD, MMSE scores were lowest in carriers of both BuChE-K and APOE ε4 and highest in carriers of BuChE-K who were APOE ε4 negative. However, a number of other studies have reported no association between BuChE-K, APOE ε4 alleles, and AD.49,53,57–61,74–76
The frequency of BuChE-K and APOE ε4 alleles in patients with dementia with Lewy bodies and Parkinson’s disease dementia has also been investigated. A higher frequency of BuChE-K and APOE ε4 was observed in patients with dementia with Lewy bodies compared with Parkinson’s disease dementia.62 Furthermore, more rapid cognitive decline was observed in patients with Parkinson’s disease dementia who were carriers of both BuChE-K and APOE ε4 compared with other genotypes.62 Singleton et al77 reported no increase in BuChE-K allele frequency between patients with dementia with Lewy bodies or Parkinson’s disease dementia; however, an increased number of BuChE-K homozygotes was observed among patients with dementia with Lewy bodies and Parkinson’s disease dementia compared with controls.77
APOE ε4 genotype has been shown to modulate BuChE phenotype, particularly in carriers of wild-type BuChE in which CSF levels of BuChE protein were approximately 30% lower in APOE ε4 carriers versus noncarriers.71 It has been hypothesized that APOE ε4 facilitates accumulation of BAβAC, that wild-type BuChE is the predominant form of BuChE protein in these complexes, and that the observed reduction in BuChE expression is a negative feedback mechanism to reduce ACh hydrolysis.71 On this basis, patients with AD and wild-type BuChE alleles may be predicted to show the greatest clinical response to BuChE inhibition.
Rationale for Inhibiting AChE and BuChE
in Patients With Alzheimer’s Disease
By inhibiting cholinesterases, cholinesterase inhibitors (ChEIs) provide a dose-dependent increase in ACh levels, enhancing cholinergic transmission in the brain of patients with AD and providing relief from symptoms of cholinergic deficits.
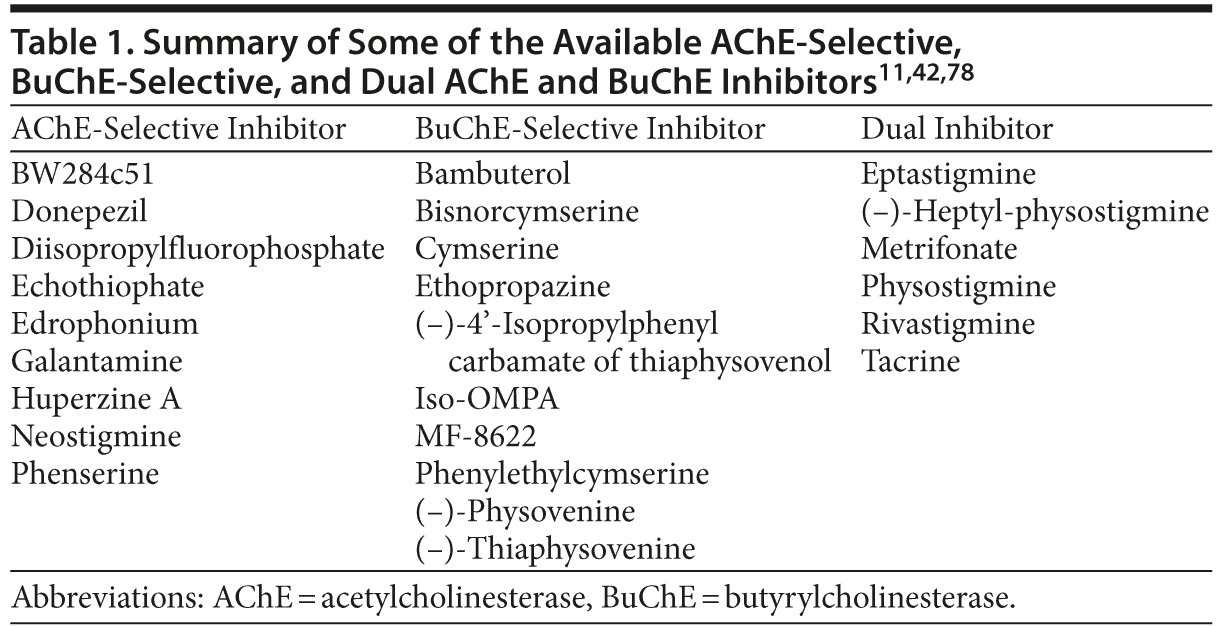
A number of ChEIs have been developed with varying potency against AChE and BuChE, many of which are close analogs of the same pharmacophore. The selectivity (AChE-selective, BuChE-selective, or dual inhibitor) of some of the available ChEIs, based on their inhibitory potency against human erythrocyte AChE and plasma BuChE, is presented in Table 1. Several of these compounds are BuChE-selective, while others (eg, rivastigmine and tacrine) show inhibitory potency against both AChE and BuChE (Table 1).
Effects of AChE- and BuChE-Selective Inhibitors: Preclinical Data
A number of preclinical studies have investigated the effect of AChE- and BuChE-selective inhibitors on the hydrolysis of ACh within the brain. AChE inhibitors may have varying potency against the different molecular forms of AChE. In the rat brain, heptylphysostigmine and diisopropylfluorophosphate showed selectivity for the G1 over the G4 form of AChE in aqueous-soluble extracts, while neostigmine was selective for G1 in both aqueous- and detergent-soluble extracts.79 Physostigmine, echothiophate, BW284c51, tacrine, and metrifonate were shown to inhibit both forms with similar potency.79 In human brain tissue, it was shown that heptylphysostigmine preferentially inhibits G1 over G4 and edrophonium inhibits G4 more potently than G1, while physostigmine inhibits both equally.41 Given that G1 is relatively unchanged in AD, while membrane-bound G4 decreases, inhibitors that preferentially inhibit the G1 form of AChE may have therapeutic applications.
Transcortical administration of donepezil, an AChE-selective inhibitor, was shown to increase extracellular ACh levels.80 Freely moving rats treated with the AChE inhibitor phenserine showed a greater than 3-fold increase in brain ACh levels.81 In 3-month-old rats with 3-(±)-(2-carboxypiperzin-4-yl)-propyl-1-phosphonic acid (CPP)–induced learning deficits, phenserine was shown to reduce the number of mistakes made in a 14-unit T maze compared with rats treated with CPP only.82
Giacobini et al11,83 reported a 15-fold increase in the extracellular concentration of ACh in rat brains perfused intracortically with MF-8622, a selective BuChE inhibitor. BuChE-selective inhibitors have also demonstrated efficacy on cognition in animal studies. Greig et al84 demonstrated that, when administered to aged rats, N1, N8-bisnorcymserine, and N1-phenethylcymserine inhibited BuChE activity, were nontoxic, and improved performance in a 14-unit T maze compared with untreated controls. The observed low cholinergic toxicity of cymserine analogs observed in the rat model is of particular interest, as centrally mediated cholinergic effects are a common barrier to treatment with high doses of ChEIs in humans.
Overview of ChEIs
ChEIs are a first-line therapy for the management of AD. Three main ChEIs are approved for the symptomatic treatment of mild-to-moderate AD: rivastigmine, galantamine, and donepezil.85–87 All 3 drugs are available as oral capsules, but rivastigmine is the only ChEI to also be approved in a transdermal patch formulation for both AD and Parkinson’s disease dementia in a number of countries worldwide.
Rivastigmine, galantamine, and donepezil are from distinct chemical classes (carbamate, phenanthrene alkaloid, and piperidine, respectively)85,86,88 and differ substantially in their pharmacologic and pharmacokinetic properties. Donepezil and galantamine are rapidly reversible inhibitors of AChE.89 Rivastigmine is a slowly reversible (pseudoirreversible) inhibitor and shows inhibitory activity against both AChE and BuChE. Under optimal assay conditions, rivastigmine has been shown to have a greater inhibitory potency (IC50) toward AChE than donepezil (4.3 nM versus 6.7 nM).90
Preclinical Data With Rivastigmine, Donepezil,
and Galantamine
Cerbai et al91 reported that intraperitoneal administration of donepezil (1 mg/kg) was associated with 27% inhibition of AChE and no inhibition of BuChE, while rivastigmine (0.6 mg/kg) inhibited AChE by 40% and BuChE by 25% in the rat cerebral cortex. Infusion of rivastigmine, but not donepezil, has been shown to increase ACh levels in the hippocampus of AChE-knockout mice, suggesting that, in the absence of AChE, rivastigmine can enhance extracellular ACh levels by inhibiting BuChE,92 and reinforcing observations that BuChE can compensate for AChE when AChE levels are depleted.
Furukawa-Hibi et al93 compared the effects of N1-phenethylcymserine, rivastigmine, and donepezil in 5-week-old imprinting control region mice with cognitive dysfunction induced by intracerebroventricular injection of Aβ peptide. Repeated daily administration of N1-phenethylcymserine, rivastigmine, and donepezil ameliorated Aβ-induced cognitive dysfunction on days 0–3 after Aβ challenge, suggesting that BuChE as well as AChE may be a therapeutic target for managing cognitive dysfunction.93
A study that utilized rivastigmine to inhibit cholinesterases in cortical plaques and tangles from persons with AD reported dose-dependent inhibition of AChE at concentrations of 10−6 to 10−4 M and complete inhibition of BuChE at 10−5 M.94 These data suggest that rivastigmine inhibits AChE and BuChE bound in plaques and tangles,94 as well as free AChE and BuChE. Furthermore, in vitro data have indicated that when cholinesterases are exposed to Aβ for several hours, AChE is inactivated by Aβ, while BuChE remains highly active.95 This finding highlights the potential importance of dual inhibition of AChE and BuChE to counteract BuChE-derived depletion of ACh in areas in which the concentration of Aβ is expected to be high. It has been suggested that rivastigmine may inhibit hyperfunctional BAβACs, as it is predicted that these complexes may be found at high concentrations in the vicinity of Aβ deposits in the brains of patients with AD. 10,94
Clinical Data Supporting the Use of ChEIs in Alzheimer’s Disease
Inhibition of AChE and BuChE in the CSF with ChEIs. In healthy volunteers, a single 3-mg dose of rivastigmine was shown to significantly inhibit AChE activity in the CSF.96 Rivastigmine treatment has also been associated with decreased AChE activity,97–101 reduced or unchanged BuChE activity,97–99,101 and decreased AChE and BuChE protein levels99 in the CSF of patients with AD. A 12-month study of rivastigmine in 11 patients with mild AD reported reductions in the activity of AChE and BuChE in both the CSF and plasma, suggesting that rivastigmine provides sustained long-term inhibition of cholinesterases.102 Giacobini et al103 reported the results of an open-label study that investigated how activity of AChE and BuChE in the CSF correlates with cognition in patients with mild-to-moderate AD receiving rivastigmine for at least 3 days. CSF levels of AChE and plasma levels of BuChE were inhibited by rivastigmine in a dose-dependent manner, and there was a significant correlation between AChE and BuChE activity and the Computerized Neuropsychological Test Battery summary score.103 Galantamine treatment has been shown to inhibit synaptic AChE variants in the CSF and the brain (measured using 1-[11C] methylpiperidin-4-yl propionate and positron emission tomography) and was associated with positive effects on the patients’ cognitive performance.104
Pharmacologic differences between the 3 commonly used ChEIs may have long-term clinical implications. Rivastigmine and donepezil have been shown to reduce AChE activity in the frontal, temporal, and parietal cortex in patients with AD.105,106 However, donepezil treatment has also been associated with increased AChE activity,97,99,101 increased BuChE activity,99 and increases in AChE and BuChE protein levels.99 Similarly, increased AChE activity has also been reported following treatment with galantamine.98,101
Significant increases in the number of cortical nicotinic ACh receptors in the brain of patients with AD have been observed following treatment with rivastigmine for 3 months, although the effect was not maintained at 12 months.107 Galantamine treatment has also been associated with sustained inhibition of AChE for up to 12 months and changes in 11C nicotine binding, which correlated positively with results on a cognitive test of attention.108 A further study demonstrated that 12 months of treatment with galantamine was associated with significant increases in regional cerebral blood flow in cortical areas, which correlated with AChE activity, nicotinic receptor activity, and cognition.109
Rivastigmine treatment has also been associated with preservation of cerebral glucose metabolism in cortical brain regions of patients with AD compared with untreated controls and a significant dose-related increase in cerebral glucose metabolism in the right frontal association region.110 A positive correlation between changes in cerebral glucose metabolism and cognitive performance was observed in patients receiving 10.5–12 mg/d of oral rivastigmine.110
Efficacy of ChEIs in patients with Alzheimer’s disease. Despite obvious pharmacologic differences, rivastigmine, galantamine, and donepezil have all demonstrated symptomatic efficacy on the ability to perform activities of daily living (ADL), behavioral symptoms, cognition, and global function in clinical trials of patients with AD.111–114
Head-to-head data comparing efficacy of rivastigmine with an AChE-selective inhibitor are limited. EXCEED (EXelon Comparison of Efficacy vErsus Donepezil) was a large, international, randomized, controlled trial that evaluated the long-term efficacy, safety, and tolerability of 3–12 mg/d of rivastigmine in capsule form and 5–10 mg/d of donepezil in 994 patients with moderate to moderately severe AD.115 Both treatment groups were comparable on measures of cognition and behavior. However, rivastigmine demonstrated superior efficacy to donepezil on the Alzheimer’s Disease Cooperative Study–ADL scale (ADCS-ADL; P = .007) and the Global Deterioration Scale (P = .049) in the intent-to-treat, last-observation-carried-forward population.115
A 6-month, open-label study of rivastigmine in 382 patients with AD who had previously failed to benefit from treatment with donepezil (due to lack of efficacy, tolerability, or both) showed that 56.2% of patients were responders to rivastigmine.116 Furthermore, a prospective, 3-month observational study investigated the efficacy of rivastigmine in patients with mild-to-moderate AD who showed deterioration when receiving treatment with an AChE-selective inhibitor.117 Switching to rivastigmine from donepezil or galantamine was associated with a response on the Clinical Global Impressions of Change in 67.7% and 66.7% of patients, respectively.117 Mean MMSE scores were also shown to improve after switching, while ADL, instrumental ADL, and Zarit scores remained stable.117 This study also reported a 30.5% reduction in the number of patients receiving concomitant antipsychotics and discontinuation of benzodiazepines in all but 1 patient following switching from an AChE-selective inhibitor to rivastigmine.117 These data provide a rationale for switching in cases of poor efficacy and tolerability. However, given the current understanding that the potential importance of BuChE as a therapeutic target increases throughout the disease course, the comparative clinical response of AChE-selective and dual AChE and BuChE inhibitors requires more detailed evaluation, particularly in patients at more advanced disease stages.
Efficacy of BuChE-selective inhibitors in patients with Alzheimer’s disease. Darvesh et al118 reported deterioration in cognitive function in a patient with AD following reduction of ethopropazine, a BuChE-selective inhibitor, and subsequent improvements following reinstatement of treatment. To date, no large-scale, randomized controlled trials of a BuChE-selective inhibitor in patients with AD have been performed. Studies in patients with mild-to-moderate AD, the patient population in the majority of ChEI trials, would be unlikely to highlight treatment advantages of BuChE inhibition.119 BuChE-selective inhibitors may be more effective than AChE-selective inhibitors in patients with severe AD, although this remains to be tested clinically.
Effect of BuChE genotype on response to ChEIs. O’Brien et al120 reported that patients with dementia with reduced-activity BuChE phenotypes show slower rates of cognitive decline and preserved attentional performance compared with patients with wild-type BuChE alleles, supporting the hypothesis that BuChE may be involved in disease progression. Patients with wild-type BuChE alleles, but not those with reduced function alleles, showed improved attention when receiving ChEI therapy.120 The potential involvement of BuChE in attention is supported by the observed distribution of BuChE in the human amygdala and hippocampal formation.16
Trials of rivastigmine provide a wealth of data pertaining to the use of dual ChEIs in the symptomatic treatment of mild-to-moderate AD. The Alzheimer’s Disease with ENA 713 (ADENA) program comprised four 26-week, randomized, double-blind trials of rivastigmine capsules in patients with mild-to-moderate AD.121–124 Stratification of patients in the ADENA database by BuChE genotype found that patients with wild-type BuChE alleles showed a significant response to rivastigmine compared with placebo on the Alzheimer’s Disease Assessment Scale–cognitive subscale (ADAS-cog; 2.29 versus 7.24, respectively; P < .0001) and Progressive Deterioration Scale (3.20 versus 9.59, respectively, P = .006). In contrast, no significant differences were observed in the response to rivastigmine in carriers of BuChE-K alleles (Novartis, data on file).
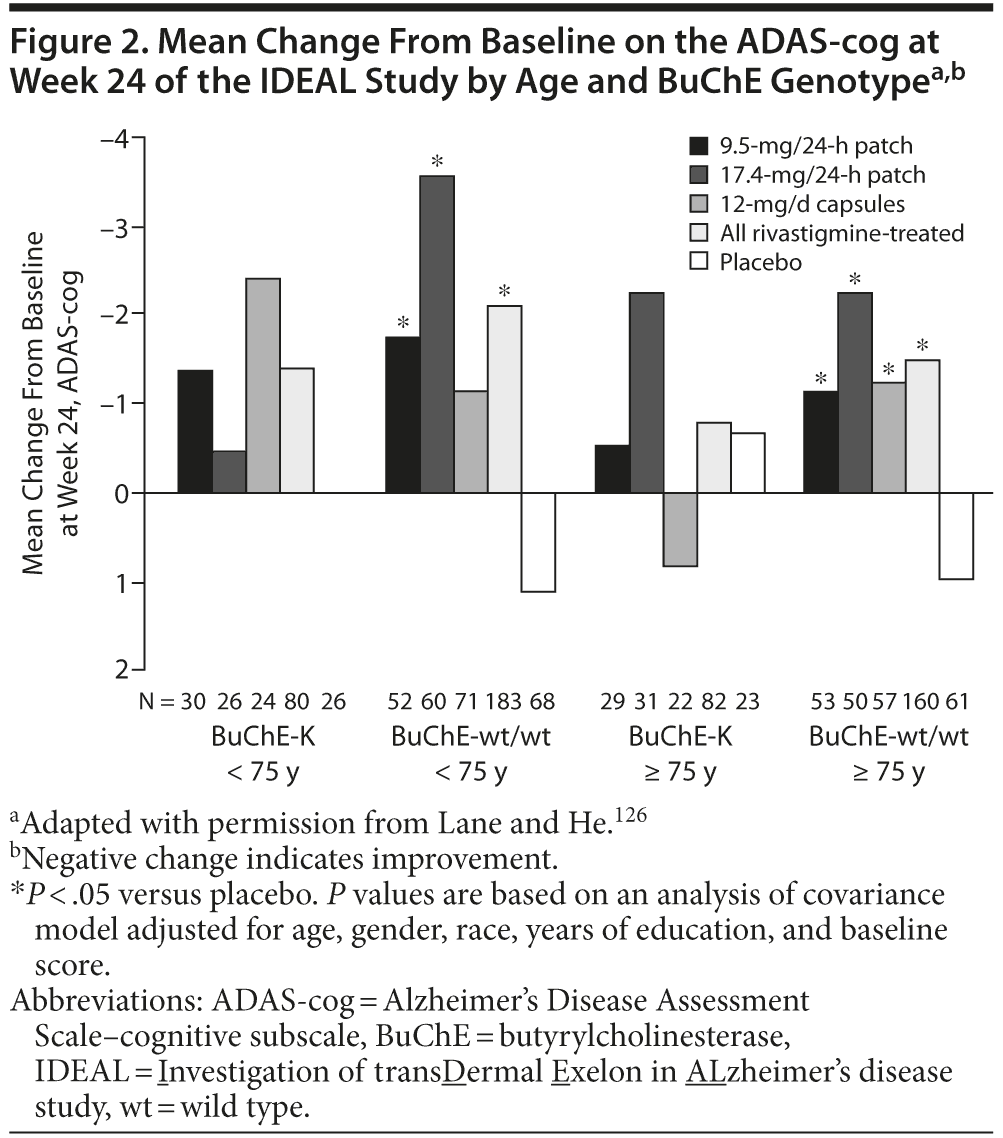
Retrospective analyses of the Investigation of transDermal Exelon in ALzheimer’s disease (IDEAL) study, a 24-week, randomized, double-blind trial of rivastigmine 9.5-mg/24-h patch, 17.4-mg/24-h patch, and 12-mg/d capsules125 have also investigated the effect of BuChE genotype and age on the response to rivastigmine. When stratified by BuChE genotype, significant differences (P < .05) were observed in the mean change from baseline on the ADAS-cog and the ADCS-ADL in rivastigmine-treated patients compared with placebo in patients with wild-type BuChE alleles but not carriers of BuChE-K (Novartis, data on file). Further stratification by age demonstrated significant changes from baseline on the ADAS-cog in rivastigmine patch–treated patients with wild-type BuChE alleles under the age of 75 years and in all rivastigmine-treated groups in patients with wild-type BuChE alleles over the age of 75 years.126 No significant effects of treatment were observed in patients with BuChE-K alleles, irrespective of age (Figure 2).126 Patients under the age of 75 years treated with 17.4-mg/24-h rivastigmine patch showed significant changes from baseline at week 24 on the ADCS-ADL.126 However, in patients over the age of 75 years, significant effects of treatment (17.4-mg/24-h patch and 12-mg/d capsule groups) were only observed in BuChE wild-type carriers and not in carriers of BuChE-K (Novartis, data on file and Lane and He126). However, it should be noted that these analyses were retrospective, were based on small numbers of patients, and were only intended to be hypothesis-forming and thus must be interpreted with caution.
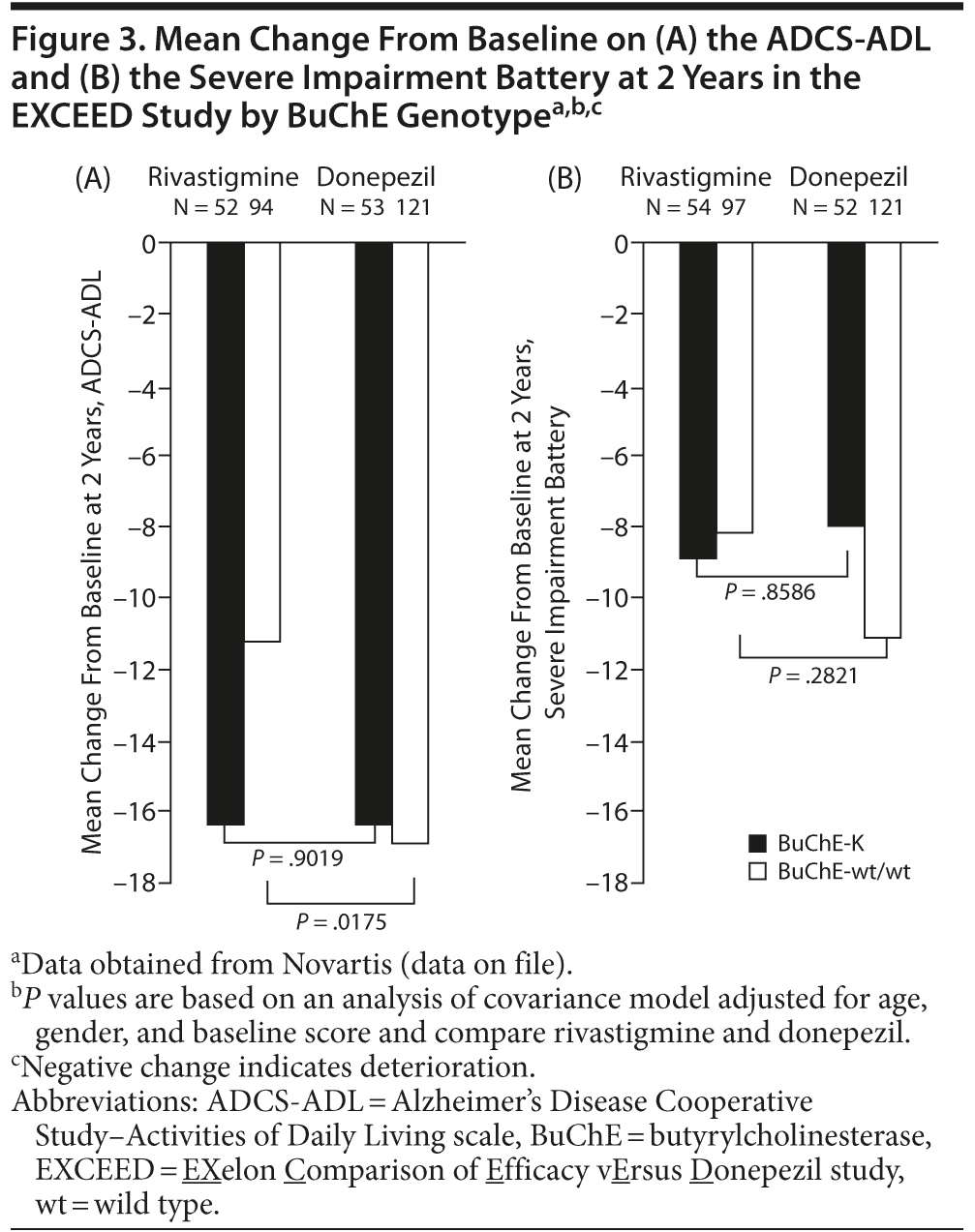
In a secondary subgroup analysis of data from the EXCEED study,115 significant differences in favor of rivastigmine compared with donepezil were observed on the Severe Impairment Battery (P = .033) and the ADCS-ADL (P = .004) in patients with wild-type BuChE and APOE ε4 genotype.
Retrospective analyses of data obtained in the EXCEED study demonstrated that patients with wild-type BuChE alleles, but not BuChE-K, showed greater response to rivastigmine than donepezil on the ADCS-ADL after 2 years of treatment (Novartis, data on file; Figure 3A). There was a trend toward greater deterioration on the Severe Impairment Battery in patients with wild-type BuChE randomized to receive donepezil compared with rivastigmine, although this did not reach significance. No significant differences in treatment response on the Severe Impairment Battery were observed in patients with BuChE-K alleles (Novartis, data on file; Figure 3B). Further retrospective analysis of data obtained in EXCEED demonstrated superiority of rivastigmine to donepezil on the ADCS-ADL and Neuropsychiatric Inventory in younger patients (< 75 years), while no significant treatment differences in favor of rivastigmine were seen in older patients.127 Treatment differences on the ADCS-ADL were particularly marked in patients with 2 wild-type BuChE alleles. Therefore, a further retrospective analysis examined the effect of BuChE genotype on the response to rivastigmine and donepezil in patients below 75 years of age.128 Younger patients with 2 wild-type BuChE alleles showed a significantly greater response to rivastigmine compared with donepezil on the ADCS-ADL and the Severe Impairment Battery. In contrast, no significant treatment differences were observed in BuChE-K carriers.128 The apparent greater efficacy of rivastigmine compared with donepezil in patients with wild-type BuChE alleles may reflect rivastigmine’s ability to inhibit both BuChE and AChE.
Overall, data from IDEAL, ADENA, and EXCEED suggest that BuChE genotype may influence outcomes and provide rationale for dual inhibition of BuChE and AChE in the management of AD. Given the nature of AD, individualized approaches to treatment may optimize therapeutic outcomes. As discussed above, genotyping for BuChE has already been used in clinical studies and has potential to be used in clinical practice; however, cost implications and time constraints limit its current clinical utility.
CONCLUSIONS
BuChE-positive neurons are found in areas of the brain involved in working memory, attention, executive function, and behavior. Mounting preclinical and clinical evidence suggests that BuChE may be important in maintaining normal cholinergic function and in neurologic conditions including AD, dementia with Lewy bodies, and Parkinson’s disease dementia and that this role may become more pronounced during the disease course. AChE-selective inhibitors and rivastigmine, a dual inhibitor of both AChE and BuChE, demonstrate efficacy in AD. Available data on the effect of BuChE genotype are limited and are largely based on prospective or retrospective analysis of clinical trial databases. These analyses were generally based on small patient numbers, and the studies were not powered to detect differences in efficacy between treatment groups stratified according to genotype. However, further investigation in more severe disease stages and advantages of dual inhibitors in specific patient populations is warranted.
An increased understanding of AD pathology, including the role of BuChE, will enable physicians to make best use of the available therapeutic options for their patients.
Drug names: donepezil (Aricept and others), edrophonium (Enlon, Tensilon, and others), galantamine (Razadyne), rivastigmine (Exelon and others), tacrine (Cognex).
Author affiliations: Alzheimer Neurobiology Center, Karolinska Institute, Stockholm, Sweden (Drs Nordberg and Darreh-Shori); Wolfson Centre for Age-Related Diseases, King’s College, London, United Kingdom (Dr Ballard); Kingshill Research Centre, Victoria Hospital, Swindon, United Kingdom (Dr Bullock); and Novartis Pharmaceuticals Corporation, East Hanover, New Jersey (Dr Somogyi).
Potential conflicts of interest: Dr Nordberg has served as a consultant to GE Healthcare and Bayer and has received honoraria from Avid, Elan, Pfizer, and Merck. Within the last 5 years, Dr Ballard has received honoraria and/or consultancy fees from Acadia, BIAL, Bristol-Myers Squibb, Eisai, Janssen, Lundbeck, Myriad, Novartis, Shire, and Servier and has received grant/research support from Acadia and Lundbeck. Dr Darreh-Shori has received honoraria from Novartis. Dr Somogyi is an employee of Novartis. Dr Bullock reports no conflicts of interest related to the subject of this study.
Funding/support: Development of this manuscript was supported by Novartis Pharmaceuticals Corporation, East Hanover, New Jersey. Novartis develops and manufactures rivastigmine and sponsored the large multicenter, randomized, double-blind trials that led to the approval of rivastigmine in the United States for mild-to-moderate dementia of the Alzheimer’s type and dementia associated with Parkinson’s disease.
Acknowledgments: Medical writing and editorial assistance in the preparation of this article were provided by Katy Cooke, PhD, of Fishawack Communications Ltd, Oxford, United Kingdom, and this service was supported by Novartis Pharmaceuticals Corporation, East Hanover, New Jersey. Dr Cooke reports no other conflicts of interest related to the subject of this article.
REFERENCES
1. Perry E, Walker M, Grace J, et al. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22(6):273–280. doi:10.1016/S0166-2236(98)01361-7 PubMed
2. Geula C. Abnormalities of neural circuitry in Alzheimer’s disease: hippocampus and cortical cholinergic innervation. Neurology. 1998; 51(suppl 1):S18–S29. PubMed
3. Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219(4589):1184–1190. doi:10.1126/science.6338589 PubMed
4. Schumacher M, Camp S, Maulet Y, et al. Primary structure of torpedo californica acetylcholinesterase deduced from its cDNA sequence. Nature. 1986;319(6052):407–409. doi:10.1038/319407a0 PubMed
5. Mesulam M, Guillozet A, Shaw P, et al. Widely spread butyrylcholinesterase can hydrolyze acetylcholine in the normal and Alzheimer brain. Neurobiol Dis. 2002;9(1):88–93. doi:10.1006/nbdi.2001.0462 PubMed
6. Adem A. Putative mechanisms of action of tacrine in Alzheimer’s disease. Acta Neurol Scand Suppl. 1992;85(S139):69–74. doi:10.1111/j.1600-0404.1992.tb04458.x PubMed
7. Daikhin Y, Yudkoff M. Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr. 2000;130(suppl 4S):1026S–1031S. PubMed
8. Messamore E, Warpman U, Ogane N, et al. Cholinesterase inhibitor effects on extracellular acetylcholine in rat cortex. Neuropharmacology. 1993;32(8):745–750. doi:10.1016/0028-3908(93)90182-3 PubMed
9. Yamada H, Otsuka M, Fujimoto K, et al. Determination of acetylcholine concentration in cerebrospinal fluid of patients with neurologic diseases. Acta Neurol Scand. 1996;93(1):76–78. doi:10.1111/j.1600-0404.1996.tb00175.x PubMed
10. Darreh-Shori T, Forsberg A, Modiri N, et al. Differential levels of apolipoprotein E and butyrylcholinesterase show strong association with pathological signs of Alzheimer’s disease in the brain in vivo. Neurobiol Aging. 2011; 32(12):2320. e15–e32. PubMed
11. Giacobini E. Selective inhibitors of butyrylcholinesterase: a valid alternative for therapy of Alzheimer’s disease? Drugs Aging. 2001;18(12):891–898. doi:10.2165/00002512-200118120-00001 PubMed
12. Oda Y, Nakanishi I. The distribution of cholinergic neurons in the human central nervous system. Histol Histopathol. 2000;15(3):825–834. PubMed
13. Brimijoin S, Hammond P. Butyrylcholinesterase in human brain and acetylcholinesterase in human plasma: trace enzymes measured by two-site immunoassay. J Neurochem. 1988;51(4):1227–1231. doi:10.1111/j.1471-4159.1988.tb03091.x PubMed
14. Green RC, Mesulam MM. Acetylcholinesterase fiber staining in the human hippocampus and parahippocampal gyrus. J Comp Neurol. 1988;273(4):488–499. doi:10.1002/cne.902730405 PubMed
15. Mesulam MM, Geula C. Acetylcholinesterase-rich neurons of the human cerebral cortex: cytoarchitectonic and ontogenetic patterns of distribution. J Comp Neurol. 1991;306(2):193–220. doi:10.1002/cne.903060202 PubMed
16. Darvesh S, Grantham DL, Hopkins DA. Distribution of butyrylcholinesterase in the human amygdala and hippocampal formation. J Comp Neurol. 1998;393(3):374–390. doi:10.1002/(SICI)1096-9861(19980413)393:3<374::AID-CNE8>3.0.CO;2-Z PubMed
17. Xie W, Stribley JA, Chatonnet A, et al. Postnatal developmental delay and supersensitivity to organophosphate in gene-targeted mice lacking acetylcholinesterase. J Pharmacol Exp Ther. 2000;293(3):896–902. PubMed
18. Li B, Stribley JA, Ticu A, et al. Abundant tissue butyrylcholinesterase and its possible function in the acetylcholinesterase knockout mouse. J Neurochem. 2000;75(3):1320–1331. doi:10.1046/j.1471-4159.2000.751320.x PubMed
19. Mesulam MM, Guillozet A, Shaw P, et al. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience. 2002;110(4):627–639. doi:10.1016/S0306-4522(01)00613-3 PubMed
20. Hartmann J, Kiewert C, Duysen EG, et al. Excessive hippocampal acetylcholine levels in acetylcholinesterase-deficient mice are moderated by butyrylcholinesterase activity. J Neurochem. 2007;100(5):1421–1429. doi:10.1111/j.1471-4159.2006.04347.x PubMed
21. Inestrosa NC, Alvarez A, Pérez CA, et al. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16(4):881–891. doi:10.1016/S0896-6273(00)80108-7 PubMed
22. Diamant S, Podoly E, Friedler A, et al. Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc Natl Acad Sci U S A. 2006;103(23):8628–8633. doi:10.1073/pnas.0602922103 PubMed
23. Das UN. Acetylcholinesterase and butyrylcholinesterase as possible markers of low-grade systemic inflammation. Med Sci Monit. 2007;13(12):RA214–RA221. PubMed
24. Perry EK, Perry RH, Blessed G, et al. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol Appl Neurobiol. 1978;4(4):273–277. doi:10.1111/j.1365-2990.1978.tb00545.x PubMed
25. Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;308(8000):1403. doi:10.1016/S0140-6736(76)91936-X PubMed
26. Whitehouse PJ, Price DL, Struble RG, et al. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215(4537):1237–1239. doi:10.1126/science.7058341 PubMed
27. Ciro A, Park J, Burkhard G, et al. Biochemical differentiation of cholinesterases from normal and Alzheimer’s disease cortex. Curr Alzheimer Res. 2012; 9(1):138–143. PubMed
28. Darreh-Shori T, Brimijoin S, Kadir A, et al. Differential CSF butyrylcholinesterase levels in Alzheimer’s disease patients with the ApoE epsilon4 allele, in relation to cognitive function and cerebral glucose metabolism. Neurobiol Dis. 2006;24(2):326–333. doi:10.1016/j.nbd.2006.07.013 PubMed
29. Geula C, Mesulam M. Special properties of cholinesterases in the cerebral cortex of Alzheimer’s disease. Brain Res. 1989;498(1):185–189. doi:10.1016/0006-8993(89)90419-8 PubMed
30. Wright CI, Geula C, Mesulam MM. Neurological cholinesterases in the normal brain and in Alzheimer’s disease: relationship to plaques, tangles, and patterns of selective vulnerability. Ann Neurol. 1993;34(3):373–384. doi:10.1002/ana.410340312 PubMed
31. Geula C, Mesulam MM. Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis Assoc Disord. 1995;9(suppl 2):23–28. doi:10.1097/00002093-199501002-00005 PubMed
32. Guillozet AL, Smiley JF, Mash DC, et al. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann Neurol. 1997;42(6):909–918. doi:10.1002/ana.410420613 PubMed
33. Mesulam MM, Geula C. Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia. Ann Neurol. 1994;36(5):722–727. doi:10.1002/ana.410360506 PubMed
34. Ruberg M, Rieger F, Villageois A, et al. Acetylcholinesterase and butyrylcholinesterase in frontal cortex and cerebrospinal fluid of demented and non-demented patients with Parkinson’s disease. Brain Res. 1986;362(1):83–91. doi:10.1016/0006-8993(86)91401-0 PubMed
35. Ruberg M, Villageois A, Bonnet AM, et al. Acetylcholinesterase and butyrylcholinesterase activity in the cerebrospinal fluid of patients with neurodegenerative diseases involving cholinergic systems. J Neurol Neurosurg Psychiatry. 1987;50(5):538–543. doi:10.1136/jnnp.50.5.538 PubMed
36. Sirviö J, Soininen HS, Kutvonen R, et al. Acetyl- and butyrylcholinesterase activity in the cerebrospinal fluid of patients with Parkinson’s disease. J Neurol Sci. 1987;81(2–3):273–279. doi:10.1016/0022-510X(87)90102-X PubMed
37. Maetzler W, Keller S, Michelis J, et al. No differences of butyrylcholinesterase protein activity and allele frequency in Lewy body diseases. Neurobiol Dis. 2009;35(2):296–301. doi:10.1016/j.nbd.2009.05.014 PubMed
38. Massoulié J, Bon S. The molecular forms of cholinesterase and acetylcholinesterase in vertebrates. Annu Rev Neurosci. 1982;5(1):57–106. doi:10.1146/annurev.ne.05.030182.000421 PubMed
39. Arendt T, Brückner MK, Lange M, et al. Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease resemble embryonic development—a study of molecular forms. Neurochem Int. 1992;21(3):381–396. doi:10.1016/0197-0186(92)90189-X PubMed
40. Siek GC, Katz LS, Fishman EB, et al. Molecular forms of acetylcholinesterase in subcortical areas of normal and Alzheimer disease brain. Biol Psychiatry. 1990;27(6):573–580. doi:10.1016/0006-3223(90)90524-6 PubMed
41. Ogane N, Giacobini E, Struble R. Differential inhibition of acetylcholinesterase molecular forms in normal and Alzheimer disease brain. Brain Res. 1992;589(2):307–312. doi:10.1016/0006-8993(92)91291-L PubMed
42. Giacobini E. Butyrylcholinesterase: its role in brain function. In: Giacobini E, ed. Butyrylcholinesterase: Its Function and Inhibitors. London, UK: Martin Dunitz; 2003:1–20.
43. Perry E, McKeith I, Ballard C. Butyrylcholinesterase and progression of cognitive deficits in dementia with Lewy bodies. Neurology. 2003;60(11):1852–1853. doi:10.1212/01.WNL.0000068336.84399.9E PubMed
44. Lehmann D, Smith A. Butyrylcholinesterase K variant in Alzheimer’s disease. In Giacobini E, ed. Butyrylcholinesterase: Its Function and Inhibitors. London, UK: Martin Dunitz; 2003: 135–148.
45. Bartels CF, Jensen FS, Lockridge O, et al. DNA mutation associated with the human butyrylcholinesterase K-variant and its linkage to the atypical variant mutation and other polymorphic sites. Am J Hum Genet. 1992;50(5):1086–1103. PubMed
46. Lane RM, He Y. Emerging hypotheses regarding the influences of butyrylcholinesterase-K variant, APOE ε 4, and hyperhomocysteinemia in neurodegenerative dementias. Med Hypotheses. 2009;73(2):230–250. doi:10.1016/j.mehy.2009.01.050 PubMed
47. Lehmann DJ, Nagy Z, Litchfield S, et al. Association of butyrylcholinesterase K variant with cholinesterase-positive neuritic plaques in the temporal cortex in late-onset Alzheimer’s disease. Hum Genet. 2000;106(4):447–452. doi:10.1007/s004390000277 PubMed
48. Ballard C, Morris C, Kalaria R, et al. The K variant of the butyrylcholinesterase gene is associated with reduced phosphorylation of tau in dementia patients. Dement Geriatr Cogn Disord. 2005;19(5-6):357–360. doi:10.1159/000084705 PubMed
49. McIlroy SP, Crawford VL, Dynan KB, et al. Butyrylcholinesterase K variant is genetically associated with late onset Alzheimer’s disease in Northern Ireland. J Med Genet. 2000;37(3):182–185. doi:10.1136/jmg.37.3.182 PubMed
50. Raygani AV, Zahrai M, Soltanzadeh A, et al. Analysis of association between butyrylcholinesterase K variant and apolipoprotein E genotypes in Alzheimer’s disease. Neurosci Lett. 2004;371(2-3):142–146. doi:10.1016/j.neulet.2004.08.057 PubMed
51. Wiebusch H, Poirier J, Sévigny P, et al. Further evidence for a synergistic association between APOE epsilon4 and BCHE-K in confirmed Alzheimer’s disease. Hum Genet. 1999;104(2):158–163. doi:10.1007/s004390050929 PubMed
52. Panegyres PK, Mamotte CD, Vasikaran SD, et al. Butyrycholinesterase K variant and Alzheimer’s disease. J Neurol. 1999;246(5):369–370. doi:10.1007/s004150050365 PubMed
53. Ghebremedhin E, Thal DR, Schultz C, et al. Age-dependent association between butyrylcholinesterase K-variant and Alzheimer disease-related neuropathology in human brains. Neurosci Lett. 2002;320(1–2):25–28. doi:10.1016/S0304-3940(02)00014-9 PubMed
54. Bizzarro A, Guglielmi V, Lomastro R, et al. BuChE K variant is decreased in Alzheimer’s disease not in fronto-temporal dementia. J Neural Transm. 2010;117(3):377–383. doi:10.1007/s00702-009-0358-y PubMed
55. Sodeyama N, Yamada M, Itoh Y, et al. Association between butyrylcholinesterase K variant and the Alzheimer type neuropathological changes in apolipoprotein E epsilon4 carriers older than 75 years. J Neurol Neurosurg Psychiatry. 1999;67(5):693–694. doi:10.1136/jnnp.67.5.693 PubMed
56. Russ C, Powell J, Lovestone S, et al. K variant of butyrycholinesterase and late-onset Alzheimer’s disease. Lancet. 1998;351(9106):881. doi:10.1016/S0140-6736(05)70292-0 PubMed
57. Singleton AB, Smith G, Gibson AM, et al. No association between the K variant of the butyrylcholinesterase gene and pathologically confirmed Alzheimer’s disease. Hum Mol Genet. 1998;7(5):937–939. doi:10.1093/hmg/7.5.937 PubMed
58. Yamamoto Y, Yasuda M, Mori E, et al. Failure to confirm a synergistic effect between the K-variant of the butyrylcholinesterase gene and the epsilon4 allele of the apolipoprotein gene in Japanese patients with Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1999;67(1):94–96. doi:10.1136/jnnp.67.1.94 PubMed
59. Kehoe PG, Williams H, Holmans P, et al. The butyrylcholinesterase K variant and susceptibility to Alzheimer’s disease. J Med Genet. 1998;35(12):1034–1035. doi:10.1136/jmg.35.12.1034 PubMed
60. Grubber JM, Saunders AM, Crane-Gatherum AR, et al. Analysis of association between Alzheimer disease and the K variant of butyrylcholinesterase (BCHE-K). Neurosci Lett. 1999;269(2):115–119. doi:10.1016/S0304-3940(99)00426-7 PubMed
61. Kim KW, Jhoo JH, Lee JH, et al. Neither the butyrylcholinesterase K variant nor transferrin C2 variant confers a risk for Alzheimer’s disease in Koreans. J Neural Transm. 2001;108(10):1159–1166. doi:10.1007/s007020170005 PubMed
62. Lane R, He Y, Morris C, et al. BuChE-K and APOE ε4 allele frequencies in Lewy body dementias, and influence of genotype and hyperhomocysteinemia on cognitive decline. Mov Disord. 2009;24(3):392–400. doi:10.1002/mds.22357 PubMed
63. Holmes C, Ballard C, Lehmann D, et al. Rate of progression of cognitive decline in Alzheimer’s disease: effect of butyrylcholinesterase K gene variation. J Neurol Neurosurg Psychiatry. 2005;76(5):640–643. doi:10.1136/jnnp.2004.039321 PubMed
64. Manoharan I, Kuznetsova A, Fisk JD, et al. Comparison of cognitive functions between people with silent and wild-type butyrylcholinesterase. J Neural Transm. 2007;114(7):939–945. doi:10.1007/s00702-007-0631-x PubMed
65. Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. PubMed
66. Darreh-Shori T, Modiri N, Blennow K, et al. The apolipoprotein E ε4 allele plays pathological roles in AD through high protein expression and interaction with butyrylcholinesterase. Neurobiol Aging. 2011;32(7):1236–1248. doi:10.1016/j.neurobiolaging.2009.07.015 PubMed
67. Mattila KM, Rinne JO, Röyttä M, et al. Dipeptidyl carboxypeptidase 1 (DCP1) and butyrylcholinesterase (BCHE) gene interactions with the apolipoprotein E epsilon4 allele as risk factors in Alzheimer’s disease and in Parkinson’s disease with coexisting Alzheimer pathology. J Med Genet. 2000;37(10):766–770. doi:10.1136/jmg.37.10.766 PubMed
68. Hiltunen M, Mannermaa A, Helisalmi S, et al. Butyrylcholinesterase K variant and apolipoprotein E4 genes do not act in synergy in Finnish late-onset Alzheimer’s disease patients. Neurosci Lett. 1998;250(1):69–71. doi:10.1016/S0304-3940(98)00453-4 PubMed
69. Lehmann DJ, Johnston C, Smith AD. Synergy between the genes for butyrylcholinesterase K variant and apolipoprotein E4 in late-onset confirmed Alzheimer’s disease. Hum Mol Genet. 1997;6(11):1933–1936. doi:10.1093/hmg/6.11.1933 PubMed
70. Tilley L, Morgan K, Grainger J, et al. Evaluation of polymorphisms in the presenilin-1 gene and the butyrylcholinesterase gene as risk factors in sporadic Alzheimer’s disease. Eur J Hum Genet. 1999;7(6):659–663. doi:10.1038/sj.ejhg.5200351 PubMed
71. Darreh-Shori T, Siawesh M, Mousavi M, et al. Apolipoprotein ε4 modulates phenotype of butyrylcholinesterase in CSF of patients with Alzheimer’s disease. J Alzheimers Dis. 2012;28(2):443–458. PubMed
72. Alvarez-Arcaya A, Combarros O, Llorca J, et al. The butyrylcholinesterase K variant is a protective factor for sporadic Alzheimer’s disease in women. Acta Neurol Scand. 2000;102(6):350–353. doi:10.1034/j.1600-0404.2000.102006350.x PubMed
73. Lane R, Feldman HH, Meyer J, et al. Synergistic effect of apolipoprotein E ε4 and butyrylcholinesterase K-variant on progression from mild cognitive impairment to Alzheimer’s disease. Pharmacogenet Genomics. 2008;18(4):289–298. doi:10.1097/FPC.0b013e3282f63f29 PubMed
74. Lee DW, Liu HC, Liu TY, et al. No association between butyrylcholinesterase K-variant and Alzheimer disease in Chinese. Am J Med Genet. 2000;96(2):167–169. doi:10.1002/(SICI)1096-8628(20000403)96:2<167::AID-AJMG8>3.0.CO;2-0 PubMed
75. Ki CS, Na DL, Kim JW, et al. No association between the genes for butyrylcholinesterase K variant and apolipoprotein E4 in late-onset Alzheimer’s disease. Am J Med Genet. 1999;88(2):113–115. doi:10.1002/(SICI)1096-8628(19990416)88:2<113::AID-AJMG2>3.0.CO;2-3 PubMed
76. Crawford F, Fallin D, Suo Z, et al. The butyrylcholinesterase gene is neither independently nor synergistically associated with late-onset AD in clinic- and community-based populations. Neurosci Lett. 1998;249(2–3):115–118. doi:10.1016/S0304-3940(98)00423-6 PubMed
77. Singleton AB, Gibson AM, Edwardson JA, et al. Butyrylcholinesterase K: an association with dementia with Lewy bodies. Lancet. 1998;351(9118):1818. doi:10.1016/S0140-6736(05)78788-2 PubMed
78. Greig NH, Sambamurti K, Yu Q-S, et al. Butyrylcholinesterase: its selective inhibition and relevance to Alzheimer’s disease therapy. In: Giacobini E, ed. Butyrylcholinesterase: Its Function And Inhibitors. London, UK: Martin Dunitz; 2003:69–90.
79. Ogane N, Giacobini E, Messamore E. Preferential inhibition of acetylcholinesterase molecular forms in rat brain. Neurochem Res. 1992;17(5):489–495. doi:10.1007/BF00969897 PubMed
80. Giacobini E, Zhu X D, Williams E, et al. The effect of the selective reversible acetylcholinesterase inhibitor E2020 on extracellular acetylcholine and biogenic amine levels in rat cortex. Neuropharmacology. 1996;35(2):205–211. doi:10.1016/0028-3908(95)00157-3 PubMed
81. Greig NH, De Micheli E, Holloway HW, et al. The experimental Alzheimer drug phenserine: preclinical pharmacokinetics and pharmacodynamics. Acta Neurol Scand suppl. 2000;102(s176):74–84. doi:10.1034/j.1600-0404.2000.00311.x PubMed
82. Patel N, Spangler EL, Greig NH, et al. Phenserine, a novel acetylcholinesterase inhibitor, attenuates impaired learning of rats in a 14-unit T-maze induced by blockade of the N-methyl-D-aspartate receptor. Neuroreport. 1998;9(1):171–176. doi:10.1097/00001756-199801050-00035 PubMed
83. Giacobini E, Griffini P, Maggi T, et al. Butyrylcholinesterase: it is important for cortical acetylcholine regulation? Soc Neurosci 1996;22:203
84. Greig NH, Utsuki T, Ingram DK, et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc Natl Acad Sci U S A. 2005;102(47):17213–17218. doi:10.1073/pnas.0508575102 PubMed
85. Galantamine (Razadyne ER) prescribing information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2004/021615lbl.pdf. Updated 2005. Accessed October 16, 2012.
86. Aricept Prescribing Information. http://www.aricept.com/assets/pdf/AriceptComboFullPINovember2010.pdf. Updated 2010. Accessed October 16, 2012.
87. Exelon US prescribing information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2006/020823s016,021025s008lbl.pdf. Updated June 2006. Accessed October 16, 2012.
88. Exelon Patch. US Prescribing Information. http://www.pharma.us.novartis.com/product/pi/pdf/exelonpatch.pdf.Updated August 2012. Accessed October 16, 2012.
89. Weinstock M. Selectivity of cholinesterase inhibition: clinical implications for the treatment of Alzheimer’s disease. CNS Drugs. 1999;12(4):307–323. doi:10.2165/00023210-199912040-00005
90. Ogura H, Kosasa T, Kuriya Y, et al. Comparison of inhibitory activities of donepezil and other cholinesterase inhibitors on acetylcholinesterase and butyrylcholinesterase in vitro. Methods Find Exp Clin Pharmacol. 2000;22(8):609–613. doi:10.1358/mf.2000.22.8.701373 PubMed
91. Cerbai F, Giovannini MG, Melani C, et al. N1phenethyl-norcymserine, a selective butyrylcholinesterase inhibitor, increases acetylcholine release in rat cerebral cortex: a comparison with donepezil and rivastigmine. Eur J Pharmacol. 2007;572(2–3):142–150. doi:10.1016/j.ejphar.2007.06.053 PubMed
92. Naik RS, Hartmann J, Kiewert C, et al. Effects of rivastigmine and donepezil on brain acetylcholine levels in acetylcholinesterase-deficient mice. J Pharm Pharm Sci. 2009;12(1):79–85. PubMed
93. Furukawa-Hibi Y, Alkam T, Nitta A, et al. Butyrylcholinesterase inhibitors ameliorate cognitive dysfunction induced by amyloid-β peptide in mice. Behav Brain Res. 2011;225(1):222–229. doi:10.1016/j.bbr.2011.07.035 PubMed
94. Eskander MF, Nagykery NG, Leung EY, et al. Rivastigmine is a potent inhibitor of acetyl- and butyrylcholinesterase in Alzheimer’s plaques and tangles. Brain Res. 2005;1060(1-2):144–152. doi:10.1016/j.brainres.2005.08.039 PubMed
95. Darreh-Shori T, Modiri N, Nordberg A. ApoE and amyloid beta deflate the cholinergic neurotransmission by boosting the activity and stability of cholinesterases in the brain. Alzheimer’s & Dementia. 2009;5(4 suppl):305.
96. Kennedy JS, Polinsky RJ, Johnson B, et al. Preferential cerebrospinal fluid acetylcholinesterase inhibition by rivastigmine in humans. J Clin Psychopharmacol. 1999;19(6):513–521. doi:10.1097/00004714-199912000-00005 PubMed
97. Amici S, Lanari A, Romani R, et al. Cerebrospinal fluid acetylcholinesterase activity after long-term treatment with donepezil and rivastigmina. Mech Ageing Dev. 2001;122(16):2057–2062. doi:10.1016/S0047-6374(01)00314-1 PubMed
98. Parnetti L, Amici S, Lanari A, et al. Cerebrospinal fluid levels of biomarkers and activity of acetylcholinesterase (AChE) and butyrylcholinesterase in AD patients before and after treatment with different AChE inhibitors. Neurol Sci. 2002;23(suppl 2):S95–S96. doi:10.1007/s100720200086 PubMed
99. Nordberg A, Darreh-Shori T, Peskind E, et al. Different cholinesterase inhibitor effects on CSF cholinesterases in Alzheimer patients. Curr Alzheimer Res. 2009;6(1):4–14. doi:10.2174/156720509787313961 PubMed
100. Cutler NR, Polinsky RJ, Sramek JJ, et al. Dose-dependent CSF acetylcholinesterase inhibition by SDZ ENA 713 in Alzheimer’s disease. Acta Neurol Scand. 1998;97(4):244–250. doi:10.1111/j.1600-0404.1998.tb00645.x PubMed
101. Parnetti L, Chiasserini D, Andreasson U, et al. Changes in CSF acetyl- and butyrylcholinesterase activity after long-term treatment with AChE inhibitors in Alzheimer’s disease. Acta Neurol Scand. 2011;124(2):122–129. doi:10.1111/j.1600-0404.2010.01435.x PubMed
102. Darreh-Shori T, Almkvist O, Guan ZZ, et al. Sustained cholinesterase inhibition in AD patients receiving rivastigmine for 12 months. Neurology. 2002;59(4):563–572. doi:10.1212/WNL.59.4.563 PubMed
103. Giacobini E, Spiegel R, Enz A, et al. Inhibition of acetyl- and butyryl-cholinesterase in the cerebrospinal fluid of patients with Alzheimer’s disease by rivastigmine: correlation with cognitive benefit. J Neural Transm. 2002;109(7–8):1053–1065. doi:10.1007/s007020200089 PubMed
104. Darreh-Shori T, Kadir A, Almkvist O, et al. Inhibition of acetylcholinesterase in CSF versus brain assessed by 11C-PMP PET in AD patients treated with galantamine. Neurobiol Aging. 2008;29(2):168–184. doi:10.1016/j.neurobiolaging.2006.09.020 PubMed
105. Kaasinen V, Någren K, Järvenpää T, et al. Regional effects of donepezil and rivastigmine on cortical acetylcholinesterase activity in Alzheimer’s disease. J Clin Psychopharmacol. 2002;22(6):615–620. doi:10.1097/00004714-200212000-00012 PubMed
106. Bohnen NI, Kaufer DI, Hendrickson R, et al. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2005;76(3):315–319. doi:10.1136/jnnp.2004.038729 PubMed
107. Kadir A, Darreh-Shori T, Almkvist O, et al. Changes in brain 11C-nicotine binding sites in patients with mild Alzheimer’s disease following rivastigmine treatment as assessed by PET. Psychopharmacology (Berl). 2007;191(4):1005–1014. doi:10.1007/s00213-007-0725-z PubMed
108. Kadir A, Darreh-Shori T, Almkvist O, et al. PET imaging of the in vivo brain acetylcholinesterase activity and nicotine binding in galantamine-treated patients with AD. Neurobiol Aging. 2008;29(8):1204–1217. doi:10.1016/j.neurobiolaging.2007.02.020 PubMed
109. Keller C, Kadir A, Forsberg A, et al. Long-term effects of galantamine treatment on brain functional activities as measured by PET in Alzheimer’s disease patients. J Alzheimers Dis. 2011;24(1):109–123. PubMed
110. Stefanova E, Wall A, Almkvist O, et al. Longitudinal PET evaluation of cerebral glucose metabolism in rivastigmine treated patients with mild Alzheimer’s disease. J Neural Transm. 2006;113(2):205–218. doi:10.1007/s00702-005-0312-6 PubMed
111. Birks J, Grimley Evans J, Iakovidou V, et al. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst Rev. 2009;(2):CD001191. PubMed
112. Loy C, Schneider L. Galantamine for Alzheimer’s disease and mild cognitive impairment. Cochrane Database Syst Rev. 2006;(1):CD001747. PubMed
113. Birks J, Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2006;(1):CD001190. PubMed
114. Birks J, Grimley Evans J, Iakovidou V, et al. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst Rev. 2000;(4):CD001191. PubMed
115. Bullock R, Touchon J, Bergman H, et al. Rivastigmine and donepezil treatment in moderate to moderately-severe Alzheimer’s disease over a 2-year period. Curr Med Res Opin. 2005;21(8):1317–1327. doi:10.1185/030079905X56565 PubMed
116. Auriacombe S, Pere JJ, Loria-Kanza Y, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease who failed to benefit from treatment with donepezil. Curr Med Res Opin. 2002;18(3):129–138. doi:10.1185/030079902125000471 PubMed
117. Bartorelli L, Giraldi C, Saccardo M, et al; Upgrade Study Group. Effects of switching from an AChE inhibitor to a dual AChE-BuChE inhibitor in patients with Alzheimer’s disease. Curr Med Res Opin. 2005;21(11):1809–1818. doi:10.1185/030079905X65655 PubMed
118. Darvesh S, MacKnight C, Rockwood K. Butyrylcholinesterase and cognitive function. Int Psychogeriatr. 2001;13(4):461–464. doi:10.1017/S1041610201007876 PubMed
119. Ballard C, Perry EK. The role of butyrylcholinesterase in Alzheimer’s disease. In: Giacobini E, ed. Butyrylcholinesterase: Its Function And Inhibitors. London, UK: Martin Dunitz; 2003: 123–134.
120. O’Brien KK, Saxby BK, Ballard CG, et al. Regulation of attention and response to therapy in dementia by butyrylcholinesterase. Pharmacogenetics. 2003;13(4):231–239. doi:10.1097/00008571-200304000-00008 PubMed
121. Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633–638. doi:10.1136/bmj.318.7184.633 PubMed
122. Corey-Bloom J, Anand R, Veach J. A randomized trial evaluating the efficacy and safety of ENA 713 (rivastigmine tartrate), a new acetylcholinesterase inhibitor, in patients with mild to moderately severe Alzheimer’s disease. Int J Geriatr Psychopharmacol. 1998;1:55–65.
123. Feldman HH, Lane R; Study 304 Group. Rivastigmine: a placebo controlled trial of twice daily and three times daily regimens in patients with Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2007;78:1056–1063. doi:10.1136/jnnp.2006.099424 PubMed
124. Schneider LS, Anand R, Farlow MR. Systematic review of the efficacy of rivastigmine for patients with Alzheimer’s disease. Int J Geriatr Psychopharmacol. 1998;1(suppl 1):S26–S34.
125. Winblad B, Cummings J, Andreasen N, et al. A six-month double-blind, randomized, placebo-controlled study of a transdermal patch in Alzheimer’s disease–rivastigmine patch versus capsule. Int J Geriatr Psychiatry. 2007;22(5):456–467. doi:10.1002/gps.1788 PubMed
126. Lane RM, He Y. Butyrylcholinesterase genotype and gender influence Alzheimer’s disease phenotype [published online ahead of print March 6, 2012]. Alzheimers Dement. 2012. doi:10.1016/j.jalz.2010.12.005 PubMed
127. Bullock R, Bergman H, Touchon J, et al. Effect of age on response to rivastigmine or donepezil in patients with Alzheimer’s disease. Curr Med Res Opin. 2006;22(3):483–494. doi:10.1185/030079906X89685 PubMed
128. Blesa R, Bullock R, He Y, et al. Effect of butyrylcholinesterase genotype on the response to rivastigmine or donepezil in younger patients with Alzheimer’s disease. Pharmacogenet Genomics. 2006;16(11):771–774. doi:10.1185/030079906X89685 PubMedc PubMed
Enjoy this premium PDF as part of your membership benefits!


