This CME activity is expired. For more CME activities, visit CMEInstitute.com.
Find more articles on this and other psychiatry and CNS topics:
The Journal of Clinical Psychiatry
The Primary Care Companion for CNS Disorders
![]()
Case Conference
The Banner Alzheimer’s Institute Case Conference is a weekly event in which physicians and staff discuss challenging and/or teaching cases of patients seen at the Institute’s Memory Disorders Clinic. These conferences are attended by a multidisciplinary group that includes Banner Alzheimer’s Institute dementia specialists, community physicians (internal medicine, family medicine, and radiology), physician assistants, social workers, nurses, medical students, residents, and fellows.
BANNER ALZHEIMER’ S INSTITUTE
The Banner Alzheimer’s Institute located in Phoenix, Arizona, has an unusually ambitious mission: to end Alzheimer’s disease without losing a generation, set a new standard of care for patients and families, and forge a model of collaboration in biomedical research. The Institute provides high-level care and treatment for patients affected by Alzheimer’s disease, dementia, and related disorders. In addition, the Institute offers extensive support services for families and many unique and rewarding research opportunities.
Prim Care Companion CNS Disord 2013;15(3):doi:10.4088/PCC.13alz01532
© Copyright 2013 Physicians Postgraduate Press, Inc.
Received: May 2, 2013; accepted May 2, 2013.
Published online: June 27, 2013.
AUTHORS
Roy Yaari, MD, MAS, a neurologist, is associate director of the Stead Family Memory Clinic of Banner Alzheimer’s Institute and a clinical professor of neurology at the College of Medicine, University of Arizona, Phoenix.
Susy Favaro, LCSW, is a clinical social worker at Banner Alzheimer’s Institute.
Anna D. Burke, MD, is a geriatric psychiatrist and dementia specialist at the Stead Family Memory Clinic of Banner Alzheimer’s Institute.
Helle Brand, PA, is a physician assistant at the Stead Family Memory Clinic of Banner Alzheimer’s Institute.
Adam S. Fleisher, MD, MAS, is associate director of Brain Imaging at Banner Alzheimer’s Institute, a neurologist at the Institute’s Stead Family Memory Clinic, and an associate professor in the Department of Neurosciences at the University of California, San Diego.
James D. Seward, PhD, ABPP, is a clinical neuropsychologist at Banner Alzheimer’s Institute.
Jan Dougherty, RN, MS, is director of Family and Community Services at Banner Alzheimer’s Institute.
Pierre N. Tariot, MD, a geriatric psychiatrist, is director of Banner Alzheimer’s Institute and a research professor of psychiatry at the College of Medicine, University of Arizona, Phoenix.
Corresponding author: Roy Yaari, MD, MAS, Banner Alzheimer’s Institute, 901 E. Willetta St, Phoenix, AZ 85006 ([email protected]).
CME Background
Original material is selected for credit designation based on an assessment of the educational needs of CME participants, with the purpose of providing readers with a curriculum of CME activities on a variety of topics from volume to volume. This special series of case reports about dementia was deemed valuable for educational purposes by the Publisher, Editor in Chief, and CME Institute Staff. Activities are planned using a process that links identified needs with desired results.
To obtain credit, read the material and go to PrimaryCareCompanion.com to complete the Posttest and Evaluation online.
CME Objective
After studying this case, you should be able to:
- Provide an appropriate workup and referral to diagnose a 57-year-old man who presents with sudden physical and cognitive decline
Accreditation Statement
The CME Institute of Physicians Postgraduate Press, Inc., is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Credit Designation
The CME Institute of Physicians Postgraduate Press, Inc., designates this journal-based CME activity for a maximum of 1.0 AMA PRA Category 1 Creditâ„¢. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Note: The American Academy of Physician Assistants (AAPA) accepts certificates of participation for educational activities certified for AMA PRA Category 1 Creditâ„¢ from organizations accredited by ACCME or a recognized state medical society. Physician assistants may receive a maximum of 1.0 hour of Category I credit for completing this program.
Date of Original Release/Review
This educational activity is eligible for AMA PRA Category 1 Creditâ„¢ through June 30, 2016. The latest review of this material was June 2013.
Financial Disclosure
All individuals in a position to influence the content of this activity were asked to complete a statement regarding all relevant personal financial relationships between themselves or their spouse/partner and any commercial interest. The CME Institute has resolved any conflicts of interest that were identified. In the past year, Larry Culpepper, MD, MPH, Editor in Chief, has been a consultant for AstraZeneca, Forest, Janssen, Lundbeck, Merck, Pfizer, and Takeda and has been a member of the speakers/advisory board for Merck. No member of the CME Institute staff reported any relevant personal financial relationships. Faculty financial disclosure appears at the end of the article.
Mr A presented to Banner Alzheimer’s Institute with his wife, who supplemented the clinical history. He was referred by a community neurologist for a second opinion.
HISTORY OF PRESENT ILLNESS (Summation of the Community Neurologist’s Evaluation Prior to Presenting to Banner Alzheimer’s Institute)
Mr A, a 57-year-old right-handed man, presented for a cognitive evaluation. Less than 1 year prior, Mr A first noted physical changes. While driving, another motorist suddenly swerved in front of him, and he had a “slow reaction speed” to brake, resulting in a minor accident without injury. Mr A stated that he had always had a very quick reaction speed, and this was unusual for him. Since that event, Mr A became more aware of subtle changes in dexterity of both his lower and upper extremities. He also developed patchy and intermittent myalgias in his legs, right arm, and neck without weakness. A cervical spine magnetic resonance image (MRI) showed no significant central canal stenosis or cord pathology, with minimal-to-mild degenerative foraminal stenosis that did not correlate with clinical symptoms. He was subsequently referred to a pain specialist. Gabapentin, hydrocodone, and neck cortisone injections provided no benefit. Mr A was unable to comply with physical therapy due to muscle cramping. He was then referred to the community neurologist.
Due to myalgias, with intermittent extremity numbness that subsequently developed, he underwent a workup for peripheral neuropathy. Electromyography and nerve conduction studies were normal with no evidence of lumbosacral radiculopathy or generalized peripheral neuropathy. Nerve biopsy, however, showed “abnormal nerve fiber density at distal sites with normal findings at proximal sites” consistent with a “length-dependent neuropathy affecting small nerve fibers.” As part of the peripheral neuropathy workup, the following tests were administered: complete blood count, comprehensive metabolic panel, thyroid-stimulating hormone, vitamin B12, folate, hemoglobin A1c, thiamine, SSA/SSB antibodies, and protein electrophoresis. All test results were normal.
Ten months after the motor vehicle accident and 2 weeks before presenting to Banner Alzheimer’s Institute, Mr A developed worsening limb dexterity that affected his gait. He also began to have difficulty with short-term memory, which appeared to worsen rapidly, and occasional involuntary limb jerks developed.
On the basis of the clinical history, what can be eliminated from the differential diagnosis?
Your Colleagues Who Attended the Banner Alzheimer’s Institute Case Conference Answered as Follows:
| A. Substance abuse | 12% |
| B. Alcohol withdrawal | 88% |
| C. Syphilis | 0% |
| D. Prion disease | 0% |
| E. Alzheimer’s disease | 6% |
| F. Encephalitis (autoimmune, bacterial, viral, or fungal) | 0% |
| G. Focal brain lesion (tumor, stroke) | 6% |
| H. Paraneoplastic syndrome | 0% |
| I. Nonconvulsive status epilepticus | 0% |
| J. Frontotemporal dementia with motor neuron disease | 0% |
| K. Multiple sclerosis | 0% |
| L. Lewy body dementia | 0% |
| M. Heavy metal/toxin exposure | 0% |
| N. All of the above should be included in the differential | 0% |
The percentages total over 100% because some conference attendees felt that more than 1 item could be eliminated from the differential diagnosis. Most attendees felt that alcohol withdrawal was not a probable cause of Mr A’s current symptoms due to the time course over many months. Others felt that his symptoms were not consistent with substance use. Due to the relatively diffuse nature of the symptoms, some believed that a focal brain lesion was not likely. Encephalitis (autoimmune, paraneoplastic, bacterial, viral, or fungal), prion disease, and Lewy body dementia were thought to be highest in the differential diagnosis.
Further testing is indicated; on the basis of the clinical history thus far, what test should not be considered?
Your Colleagues Who Attended the Banner Alzheimer’s Institute Case Conference Answered as Follows:
| A. Lumbar puncture (cell count, glucose, protein, 14-3-3 protein, fungal screen, viral screen, bacterial) | 0% |
| B. MRI brain | 0% |
| C. FDG-PET brain | 100% |
| D. EEG | 0% |
| E. All tests are indicated | 0% |
All conference attendees believed that lumbar puncture, MRI, and EEG would be useful tests and that the FDG-PET was not indicated currently. The clinical indication for FDG-PET is to help distinguish between Alzheimer’s disease and frontotemporal dementia, neither of which is very likely. Furthermore, a structural brain scan (either head computerized tomography or MRI) should be conducted as a first step prior to consideration of an FDG-PET.
It should be noted that a structural brain scan should also be conducted prior to lumbar puncture to rule out a mass lesion or cerebral edema. A lumbar puncture should not be performed in a patient with increased intracranial pressure due to a space-occupying lesion or cerebral edema because the sudden release of cerebrospinal fluid (CSF) can lower the pressure in the spinal column, resulting in brain herniation through the foramen magnum (Bradley et al, 1999).
Neuropsychological testing was suggested as an option should the cognitive process require better characterization for diagnostic purposes. It was agreed that the EEG, MRI, and lumbar puncture should be done first, and, then, if further information is necessary, neuropsychological testing should be considered.
TEST RESULTS
All basic blood test results were unremarkable, including the erythrocyte sedimentation rate test. An EEG showed localized sharp waves intermittently in the left temporal area, as well as focal slowing in the left temporal region. Routine CSF studies, including cell count, were normal. Fungal and viral tests were normal. Because of the physical and cognitive changes, an MRI of the brain was ordered by Mr A’s treating neurologist.
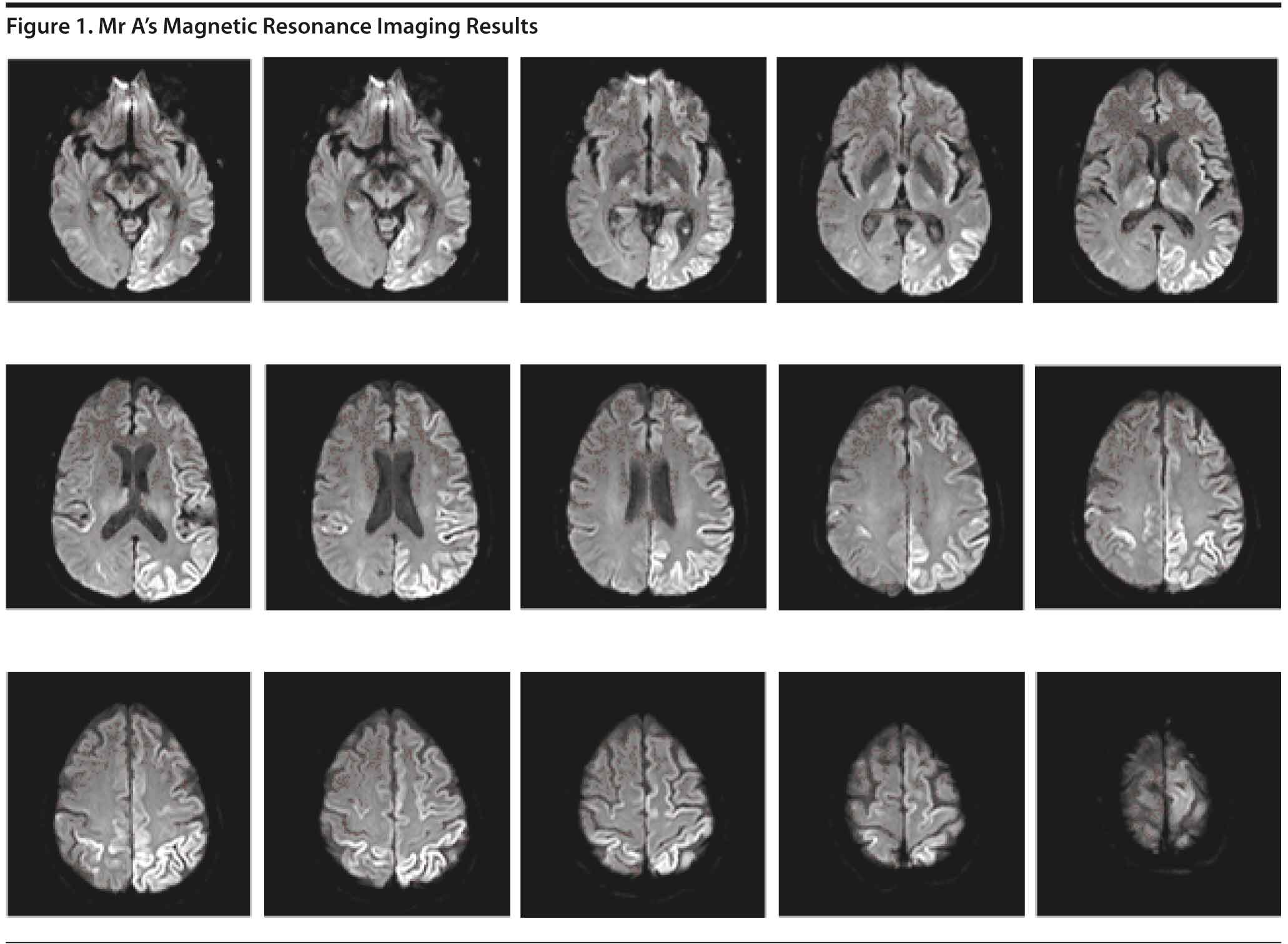
The MRI (Figure 1) revealed increased diffusion-weighted imaging signal in the cortex of the left parietal and occipital lobes with some scattered bilateral frontal lobe involvement, right parietal involvement, and scattered bilateral thalamic involvement. Due to the abnormal MRI findings, a lumbar puncture was subsequently performed for diagnostic purposes. The CSF results showed no evidence of infection, inflammation, or hemorrhage.
HISTORY OF PRESENT ILLNESS (Presentation to Banner Alzheimer’s Institute for a Second Opinion Approximately 10 Months After Symptom Onset)
When Mr A presented to Banner Alzheimer’s Institute, his wife noted that, over the past 2 weeks, he had experienced a significant functional decline both physically and cognitively. He had required use of a walker for ambulation for 2 days and had a fall from which he was unable to arise unassisted. His short-term memory had rapidly declined, and he was let go from his work as a machinist approximately 2 weeks before. Work performance had rapidly declined for about a month, which had never been an issue before. Mr A had experienced no significant personality changes, but his wife noted a recent argument over a minor financial issue, which was out of character for him and resolved quickly. He did have some minor paranoia, which had been worsening, such as wanting the curtains closed at night, with some suspiciousness of others. There were no visual hallucinations or delusions. Since his rather sudden physical and cognitive decline over the past 2 weeks, Mr A had been depressed and “into himself,” not talking very much. He had no prior history of depression.
At the time of the visit, Mr A was not repetitive but was no longer able to track appointments. He misplaced items, and word-finding difficulty affected his speech. He was not oriented to the day of the week. He was able to recognize friends and family but unable to perform personal finances. His wife had asked him to stop driving 1 week before due to safety concerns. Within the past week, he had begun to require assistance with medication management. The apraxia worsened. He had trouble using a telephone and developed worsened myoclonus within the past month. Mr A had cooked a meal 3 weeks before without difficulty, but his wife did not believe he would be able to do so now due to “dexterity.” He had stopped using the computer recently, including social networks, which he previously enjoyed. He required minor assistance with showering and dressing due to physical limitations. Toileting was intact. He had recent insomnia, which was improved on his current medication regimen.
PAST MEDICAL HISTORY
Mr A had a history of hypertension. He was involved in a motorcycle accident with no helmet in April 2007, with a fracture to his knee, wrist, and clavicle and a blow to his head with a brief loss of consciousness. There were no cognitive sequelae at that time. Mr A also was involved in a car accident 1 year ago, as mentioned previously, with an injury to his patella.
ALLERGIES
Mr A had no known drug allergies.
MEDICATIONS
Mr A’s medications included hydrocodone/acetaminophen 5/500 mg, which he had been taking 3-4 times daily for pain for the past 10 months; losartan; carbamazepine 200 mg twice daily for tremor control; clonazepam 0.5 mg at bedtime, also to help with tremors; gabapentin 900 mg three times daily to help with neuropathic pain; and tizanidine, which was recently added at 4 mg twice daily.
SOCIAL HISTORY
Mr A had 12 years of education and worked as a machinist until he was let go. He lived with his wife. Mr A had no history of chronic alcohol abuse and quit smoking 10-15 years ago. There was no history of illicit substance use.
FAMILY HISTORY
Mr A’s mother was in her mid-70s and had a “touch of subtle forgetfulness.” There was no history of any known neurologic or psychiatric disorders in the family.
REVIEW OF SYSTEMS
A complete review of systems provided no additional information.
PHYSICAL EXAMINATION
Mr A’s vitals included blood pressure: 140/88, pulse: 88 bpm, weight: 187 lb, and height: 64 in. His general physical examination was unremarkable except for mild and subtle bilateral cataracts.
NEUROLOGIC EXAMINATION
The neurologic examination was remarkable for the following findings. Eye movements demonstrated broken smooth pursuits. Coordination testing showed arrhythmia with decreased amplitude bilaterally, worse on the right than on the left. Mr A had significant right arm apraxia. Deep tendon reflexes were brisk but symmetrical throughout. He had a positive glabellar and snout reflex and an occasional myoclonic jerk. His gait was unsteady, and he required the use of a walker. Mr A demonstrated increased tone throughout, right side greater than the left. Deep tendon reflexes were brisk, but symmetric, with bilateral normal plantar reflex.
A Mini-Mental State Examination score generally correlates with disease severity. Scores ≤ 9 points can indicate severe dementia, scores between 10-20 points can indicate moderate dementia, and scores > 20 points can indicate mild dementia (Mungas, 1991). Although MMSE scores must be interpreted in light of both the patient’s age and education, education is the primary demographic factor that affects scores. Therefore, whereas a cutoff of ≤ 23 is widely used in distinguishing between normal and abnormal performance, this cutoff may have less predictive ability in poorly educated individuals (Folstein et al, 1975).
On the basis of the information presented thus far, what would you expect the MMSE score to be?
Your Colleagues Who Attended the Banner Alzheimer’s Institute Case Conference Answered as Follows:
| A. 26-30 | 0% |
| B. 21-25 | 33% |
| C. 16-20 | 67% |
| D. 11-15 | 0% |
| E. < 11 | 0% |
Figure 2 shows Mr A’s pentagon drawing and sentence from the MMSE. On the basis of the clinical history, most conference attendees felt that Mr A would score in the mild-to-moderate range. Mr A scored 15 points on the MMSE.

The Montreal Cognitive Assessment (MoCA) is a 30-point test that assesses several cognitive domains. Because it is more challenging than the Mini-Mental State Examination, it has greater sensitivity for mild cognitive impairment and early stages of dementia. With a cutoff score < 26, the sensitivity for detecting mild cognitive impairment (N = 94) was 90% and the specificity was 87% (Nasreddine et al, 2005). Research has demonstrated that MoCA scores are highly correlated with education. It is recommended that education be taken into account when interpreting MoCA performance, but there are no formal specific cutoff scores for lower education at this time (Johns et al, 2008). This test is available online at http://mocatest.org/.
On the basis of the information presented thus far, what would you expect the MoCA score to be?
Your Colleagues Who Attended the Banner Alzheimer’s Institute Case Conference Answered as Follows:
| A. 26-30 | 0% |
| B. 21-25 | 0% |
| C. 16-20 | 14% |
| D. 11-15 | 64% |
| E. < 11 | 22% |
Figure 3 shows Mr A’s clock drawing. He scored 12 points on the MoCA (Figure 4). Figure 5 shows Mr A’s Category Retrieval Test results.



The DSM-IV defines dementia as multiple cognitive deficits that include memory impairment and at least 1 of the following cognitive disturbances: aphasia, apraxia, agnosia, or a disturbance in executive functioning. The cognitive deficits must be sufficiently severe to cause impairment in social or occupational functioning and must represent a decline from a previously higher level of functioning. A diagnosis of dementia should not be made if the cognitive deficits occur exclusively during the course of a delirium (American Psychiatric Association, 2000).
On the basis of the information presented thus far, do you believe that the patient meets criteria for dementia?
Your Colleagues Who Attended the Banner Alzheimer’s Institute Case Conference Answered as Follows:
| A. Yes | 100% |
| B. No | 0% |
Given the workup thus far, all conference attendees believed that a rapidly progressive dementia was present.
On the basis of the information presented thus far, what is the most likely diagnosis?
Your Colleagues Who Attended the Banner Alzheimer’s Institute Case Conference Answered as Follows:
| A. Frontotemporal dementia | 0% |
| B. Alzheimer’s disease | 0% |
| C. Creutzfeldt-Jakob disease | 100% |
| D. Corticobasal ganglionic degeneration | 0% |
| E. Multiple system atrophy | 0% |
All conference attendees believed that the clinical profile in conjunction with the MRI and other test results were consistent with Creutzfeldt-Jakob disease.
What further test is indicated?
Your Colleagues Who Attended the Banner Alzheimer’s Institute Case Conference Answered as Follows:
| A. Amyloid brain imaging | 0% |
| B. Neuropsychological testing | 0% |
| C. Repeat MRI due to possible artifact on prior test | 0% |
| D. No further tests | 100% |
At this time, given the high likelihood of Creutzfeldt-Jakob disease, all attendees were in agreement that no further tests were indicated. A 14-3-3 CSF protein test was pending, which was ordered by Mr A’s primary neurologist at the time of the lumbar puncture.
The 14-3-3 protein in cerebrospinal fluid is a known biomarker for Creutzfeldt-Jakob disease but, unfortunately, has limited sensitivity. In a cohort of 32 pathologically confirmed cases, only 17 (53%) showed elevated cerebrospinal fluid 14-3-3 protein levels (Geschwind et al, 2003).
IMPRESSION OF THE TREATING PHYSICIAN AT FIRST VISIT to Banner Alzheimer’s Institute
Mr A is a 57-year-old right-handed man who presented for a cognitive evaluation. The patient’s clinical history and cognitive findings are consistent with a rapidly progressing dementia coupled with apraxia and myoclonus. Test results, especially the brain MRI, are consistent with Creutzfeldt-Jakob disease. A 14-3-3 CSF protein test is pending. The patient’s family needs support.
PLAN
- No further testing is recommended.
- Await the results of the 14-3-3 CSF protein test.
- Suggest minimizing the medication regimen as permitted, especially tizanidine, which could further worsen Mr A’s cognition.
- Cholinesterase inhibitors or memantine are not indicated.
- Mr A’s wife was referred to meet with a social worker with the Family and Community Services team for individual psychotherapy for adjustment to her husband’s illness as well as grief support.
- Mr A and his wife were referred to the National Prion Disease Pathology Surveillance Center (http://www.cjdsurveillance.com/) in order to help establish the diagnosis of prion disease through autopsy.
- Hospice care was advised due to the poor prognosis. Mr A’s wife was advised to expect continued rapid cognitive and physical deterioration with death within months. A specific timeframe could not be given, but it was noted that death typically occurs within 1 year of symptom onset (Haywood, 1997)
- His wife was reassured that Creutzfeldt-Jakob disease cannot be transmitted through the air or casual contact, and spouses of patients have no higher risk of contracting the disease. It was explained that the disease can be transmissible via exposure to brain tissue and spinal cord fluid from infected individuals.
- Mr A is to follow up with the treating physician on an as-needed basis and continue to be seen by his primary neurologist as needed.
Two weeks Later: Social Worker Appointment With Mr A’s Wife at Banner Alzheimer’s Institute
Mr A’s wife reported continued rapid decline in his functional and cognitive status. He had little to no use of his right upper extremity. He continued to ambulate with a walker but was less stable and easily fatigued. He required assistance for personal activities of daily life, such as bathing and dressing. He could no longer use utensils and ate “finger foods.” He talked less, sometimes using only a few words. His wife was uncertain as to his level of comprehension.
Mr A’s wife had recently begun the process of applying for Social Security disability and contacted his employer regarding disability insurance. Medical power of attorney forms were completed, but mental health (required in Arizona for psychiatric hospitalization) or financial powers of attorney were not. These forms were provided to her.
Relatives had arrived to spend time with Mr A and assist his wife, as caring for him had become “overwhelming.” His wife expressed commitment to doing what she could to make his life as good as possible.
Communication strategies to accommodate changes noted by Mr A’s wife were reviewed, including not quizzing him or asking him what he wanted, in that it appeared that choosing options was too overwhelming for him. Instead, she was to offer food and drink throughout the day as she suspected would be comforting to him. Activity options were to focus on sensory stimulation, such as hand massages with lotion, back rubs, music, reading aloud, and movies and TV that featured light comedy and upbeat subject matter. Mr A’s wife contacted hospice and was visited the next day for an initial assessment.
FIVE weeks After Mr A’s Initial Appointment at Banner Alzheimer’s Institute (About 1 Year Since Symptom Onset)
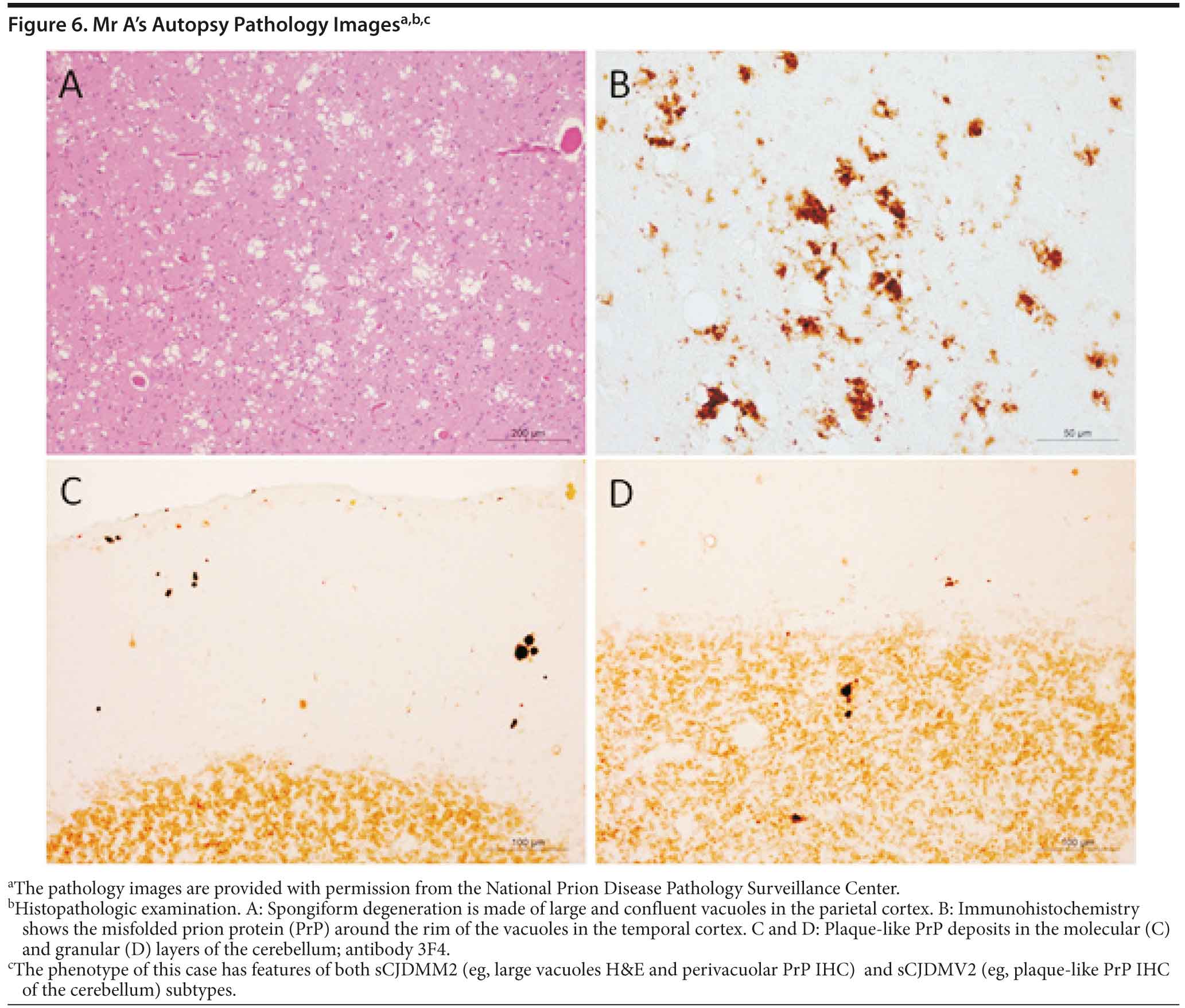
Mr A died. A brain autopsy was performed, and immunostaining was consistent with prion disease, specifically sporadic Creutzfeldt-Jakob disease (nonfamilial) (Figure 6). The 14-3-3 CSF protein test was negative.
DISCUSSION
Prion diseases, referred to as transmissible spongiform encephalopathies, are a family of rare progressive neurodegenerative disorders that occur in humans and animals. Brain autopsy of patients with prion disease reveals vacuolization of the brain tissue, called spongiform degeneration (meaning that the tissue deteriorates, developing a spongy texture). Also present in affected brain tissue is an abnormal protein called scrapie prion protein (PrPSc). The PrPSc is believed to result from a change in the conformation or shape of a normal protein called cellular prion protein (PrPC). Since the abnormal prion protein cannot be broken down through the body’s normal process, it aggregates mostly in the brain, causing degeneration and disease (http://www.cjdsurveillance.com/abouthpd.html).
Animal prion disease includes bovine spongiform encephalopathy, otherwise known as mad cow disease, and scrapie, which affects sheep and goats. The most common human prion disease is Creutzfeldt-Jakob disease. There are 3 types of Creutzfeldt-Jakob disease: (1) sporadic, also called spontaneous, for which the cause is not known; (2) familial, also called genetic or inherited, which is due to a defect in the prion protein gene; and (3) acquired (referred to as variant), which is transmitted by infection due to exposure to the infectious prion from contaminated meat or from transplant of contaminated tissues or use of contaminated instruments during surgical procedures.
Sporadic Creutzfeldt-Jakob disease, although rare (incidence of 1 per 1-2 million/year), is the most common variation, accounting for 85% of patients with the disease (Miller et al, 2009). The disease presents between ages 50 and 70 years and has a median survival of 5 months, with an 85% death rate within the first year of symptom onset (Geshwind et al, 2008). Iatrogenic Creutzfeldt-Jakob disease has been reported in patients who have received cadaveric growth hormone, pituitary hormone, and human gonadotropin. Other cases of iatrogenic Creutzfeldt-Jakob disease have been reported from a dura mater graft, a corneal graft, and exposure to neurosurgical equipment that has been exposed to prion (Haywood, 1997). Given the lack of such history in Mr A, iatrogenic Creutzfeldt-Jakob disease was not a consideration. Acquired (or new variant) Creutzfeldt-Jakob disease was also not a likely etiology in this case. New variant Creutzfeldt-Jakob disease, thought to be acquired from eating neural tissue of animals infected with prion disease, is less common, presents at a younger age (median age of about 29 years) with predominantly behavioral changes, and has a slightly longer time course (Haywood, 1997). Mr A had not travelled to areas endemic to bovine spongiform encephalopathy.
According to the World Health Organization, patients usually experience psychiatric or sensory symptoms that most commonly take the form of depression, apathy, or anxiety and, occasionally (in a third of the cases), unusual persistent and painful sensory symptoms. Paresthesias can occur when the thalamus is affected, as was the case in Mr A. Neurologic signs, including unsteadiness, difficulty walking, and involuntary movements, develop as the illness progresses, and, by the time of death, patients become completely immobile and mute (http://www.who.int/mediacentre/factsheets/fs180/en/). Because of this highly variable clinical presentation that includes cognitive, behavioral, sensory, or motor dysfunction, Creutzfeldt-Jakob disease can be very difficult to diagnose, with approximately 15%-20% of suspected Creutzfeldt-Jakob disease cases resulting in misdiagnosis (Geschwind et al, 2007).
The workup for suspected Creutzfeldt-Jakob disease includes spinal fluid analysis, EEG, and brain MRI. The typical CSF profile in a patient with Creutzfeldt-Jakob disease shows a mildly elevated protein and normal glucose without leukocytosis (Geschwind et al, 2008). If signs of an inflammatory process are present, an autoimmune-mediated encephalopathy should be considered (Rosenbloom and Atri, 2011). The 14-3-3 protein in CSF is a known biomarker for Creutzfeldt-Jakob disease but, unfortunately, has limited sensitivity. In a cohort of 32 pathologically confirmed cases, only 17 (53%) showed elevated 14-3-3 CSF protein levels (Geschwind et al, 2003).
The EEG is useful in detecting cortical irritability in Creutzfeldt-Jakob disease. In the early stages of the disease, patients may show focal or diffuse slowing, and, in later stages, the characteristic 1-2 Hz periodic sharp waves may be present. These EEG findings, however, are not specific to Creutzfeldt-Jakob disease and have limited sensitivity and specificity (Geschwind et al, 2008).
Brain MRI can be a useful diagnostic tool for Creutzfeldt-Jakob disease. The typical MRI findings in a patient with sporadic Creutzfeldt-Jakob disease include fluid attenuated inversion recovery and diffusion-weighted imaging hyperintensities involving cortical and subcortical gray matter (thalamus, striatum) (Rosenbloom and Atri, 2011).
The appropriate workup is very important in suspected Creutzfeldt-Jakob disease, especially because there are reversible and treatable forms of “rapidly progressive dementia,” including limbic encephalitis and Hashimoto’s encephalopathy. For a comprehensive review on the evaluation of a patient with rapidly progressive dementia, the articles by Geschwind et al (2010) and Rosenblum and Atri (2011) are recommended.
Disclosure of off-label usage
The authors have determined that, to the best of their knowledge, no investigational information about pharmaceutical agents that is outside US Food and Drug Administration-approved labeling has been presented in this activity.
FINANCIAL DISCLOSURE
Dr Yaari is a consultant for Amedisys Home Health. Dr Tariot has served as a consultant for Abbott, AC Immune, Adamas, Avanir, Boehringer-Ingelheim, Chase, Chiesi, Eisai, Elan, MedAvante, Merz, Neuroptix, Otsuka, and Sanofi-Aventis; has received consulting fees and research support from AstraZeneca, Avid, Bristol-Myers Squibb, Eli Lilly, Genentech, GlaxoSmithKline, Janssen, Medivation, Merck, Pfizer, Roche, and Toyama; has received research support only from Baxter, Functional Neuromodulation, GE, and Targacept; has received other research support from Alzheimer’s Association, Arizona Department of Health Services, National Institute of Mental Health, and National Institute on Aging; is a stock shareholder in Adamas; and is listed as a contributor to a patent owned by the University of Rochester (Rochester, New York) for “Biomarkers of Alzheimer’s Disease.” Drs Burke, Fleisher, and Seward and Mss Favaro, Brand, and Dougherty have no personal affiliations or financial relationships with any commercial interest to disclose relative to the activity.
FUNDING/SUPPORT
None reported.
ACKNOWLEDGMENTS
The authors would like to acknowledge the National Prion Disease Pathology Surveillance Center (Cleveland, Ohio) for their assistance with this patient and Ignazio Cali, MS (National Prion Disease Pathology Surveillance Center, Department of Pathology, Case Western University, Cleveland, Ohio), for the pathology images. Mr Cali reports no conflicts of interest related to the subject of this article.
DISCLAIMER
The opinions expressed are those of the authors, not of Banner Health or Physicians Postgraduate Press.
Clinical Points
- The work-up of a rapidly progressive dementia includes structural brain imaging, spinal fluid analysis, and electroencephalogram.
- Clinical symptoms that indicate Creutzfeldt-Jakob disease include depression, apathy, anxiety, pain, and sometimes paresthesia; progression of the illness leads to motor dysfunction and finally to immobilization and muteness.
- Provide counseling to family members of a patient diagnosed with Creutzfeldt-Jakob disease.
Find more articles on this and other psychiatry and CNS topics:
The Journal of Clinical Psychiatry
The Primary Care Companion for CNS Disorders
REFERENCES
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. Arlington, VA: American Psychiatric Association; 2000.
Bradley WG, Daroff RB, Marsden CD, et al, eds. Neurology in Clinical Practice: Principles of Diagnosis and Management, 3rd ed. Burlington, MA: Butterworth-Heinemann; 1999:467.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198. PubMed doi:10.1016/0022-3956(75)90026-6
Geschwind MD, Haman A, Miller BL. Rapidly progressive dementia. Neurol Clin. 2007;25(3):783-807, vii. 1. PubMed doi:10.1016/j.ncl.2007.04.001
Geschwind MD, Martindale J, Miller D, et al. Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003;60(6):813-816. PubMed doi:10.1001/archneur.60.6.813
Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64(1):97-108. PubMed doi:10.1002/ana.21430
Geschwind MD. Rapidly progressive dementia: prion diseases and other rapid dementias. Continuum (Minneap Minn). 2010;16(2 Dementia):31-56. PubMed
Haywood AM. Transmissible spongiform encephalopathies. N Engl J Med. 1997;337:1821-1828. doi:10.1056/NEJM199712183372508 PubMed
Johns EK, Phillips NA, Chertkow H, et al. The effect of education on performance on the Montreal Cognitive Assessment (MoCA): normative data from the community [poster]. Canadian J Geriatrics. 2008:11(1):62. Presented at the 28th Annual Meeting of the Canadian Geriatrics Society; April 2008; Montreal, Quebec, Canada
Johns EK, Phillips NA, Chertkow H, et al. The Montreal Cognitive Assessment: normative data in the community. In: Final Program of the 36th Annual Meeting of the International Neuropsychological Society; February 6-9, 2008; Waikoloa, Hawaii. J Int Neuropsychol Soc. 2008;41(suppl 1):58.
Miller B, Bave B, eds. The Behavioral Neurology of Dementia. 1st ed. Cambridge, UK: Cambridge University Press; 2009. doi:10.1017/CBO9780511581410
Mungas D. In-office mental status testing: a practical guide. Geriatrics. 1991;46(7):54-58, 63, 66. PubMed
Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695-699. PubMed doi:10.1111/j.1532-5415.2005.53221.x
Rosenbloom MH, Atri A. The evaluation of rapidly progressive dementia. Neurologist. 2011;17(2):67-74. PubMed doi:10.1097/NRL.0b013e31820ba5e3
Enjoy this premium PDF as part of your membership benefits!



