See Commentary by Tohen
Article Abstract
Objective: This phase 3 trial evaluated the efficacy, safety, and tolerability of low- and high-dose cariprazine in patients meeting DSM-IV-TR criteria for acute manic or mixed episodes associated with bipolar I disorder.
Method: This multicenter, randomized, double-blind, placebo-controlled, parallel-group, fixed/flexible-dose study was conducted from February 2010 to December 2011. Patients were randomly assigned to placebo, cariprazine 3-6 mg/d, or cariprazine 6-12 mg/d for 3 weeks of double-blind treatment. Primary and secondary efficacy parameters were change from baseline to week 3 in Young Mania Rating Scale (YMRS) total score and Clinical Global Impressions-Severity of Illness (CGI-S) score, respectively. Post hoc analysis examined change from baseline to week 3 in YMRS single items.
Results: A total of 497 patients were randomized; 74% completed the study. The least squares mean difference (LSMD) for change from baseline to week 3 in YMRS total score was statistically significant in favor of both cariprazine groups versus placebo (LSMD [95% CI]: 3-6 mg/d, −6.1 [−8.4 to −3.8]; 6-12 mg/d, −5.9 [−8.2, −3.6]; P < .001 [both]). Both cariprazine treatment groups showed statistically significant superiority to placebo on all 11 YMRS single items (all comparisons, P < .05). Change from baseline in CGI-S scores was statistically significantly greater in both cariprazine groups compared with placebo (LSMD [95% CI]: 3-6 mg/d, −0.6 [−0.9 to −0.4]; 6-12 mg/d, −0.6 [−0.9 to −0.3]; P < .001 [both]). The most common (≥ 5% and twice the rate of placebo) treatment-related adverse events for cariprazine were akathisia (both groups) and nausea, constipation, and tremor (6-12 mg/d only).
Conclusions: Results of this study demonstrated that both low- and high-dose cariprazine were more effective than placebo in the treatment of acute manic or mixed episodes associated with bipolar I disorder. Cariprazine was generally well tolerated, although the incidence of akathisia was greater with cariprazine than with placebo.
Trial Registration: ClinicalTrials.gov identifier: NCT01058668
This work may not be copied, distributed, displayed, published, reproduced, transmitted, modified, posted, sold, licensed, or used for commercial purposes. By downloading this file, you are agreeing to the publisher’s Terms & Conditions.
Efficacy and Safety of Low- and High-Dose Cariprazine in Acute and Mixed Mania Associated With Bipolar I Disorder: A Double-Blind, Placebo-Controlled Study

ABSTRACT
Objective: This phase 3 trial evaluated the efficacy, safety, and tolerability of low- and high-dose cariprazine in patients meeting DSM-IV-TR criteria for acute manic or mixed episodes associated with bipolar I disorder.
Method: This multicenter, randomized, double-blind, placebo-controlled, parallel-group, fixed/flexible-dose study was conducted from February 2010 to December 2011. Patients were randomly assigned to placebo, cariprazine 3-6 mg/d, or cariprazine 6-12 mg/d for 3 weeks of double-blind treatment. Primary and secondary efficacy parameters were change from baseline to week 3 in Young Mania Rating Scale (YMRS) total score and Clinical Global Impressions-Severity of Illness (CGI-S) score, respectively. Post hoc analysis examined change from baseline to week 3 in YMRS single items.
Results: A total of 497 patients were randomized; 74% completed the study. The least squares mean difference (LSMD) for change from baseline to week 3 in YMRS total score was statistically significant in favor of both cariprazine groups versus placebo (LSMD [95% CI]: 3-6 mg/d, −6.1 [−8.4 to −3.8]; 6-12 mg/d, −5.9 [−8.2, −3.6]; P < .001 [both]). Both cariprazine treatment groups showed statistically significant superiority to placebo on all 11 YMRS single items (all comparisons, P < .05). Change from baseline in CGI-S scores was statistically significantly greater in both cariprazine groups compared with placebo (LSMD [95% CI]: 3-6 mg/d, −0.6 [−0.9 to −0.4]; 6-12 mg/d, −0.6 [−0.9 to −0.3]; P < .001 [both]). The most common (≥ 5% and twice the rate of placebo) treatment-related adverse events for cariprazine were akathisia (both groups) and nausea, constipation, and tremor (6-12 mg/d only).
Conclusions: Results of this study demonstrated that both low- and high-dose cariprazine were more effective than placebo in the treatment of acute manic or mixed episodes associated with bipolar I disorder. Cariprazine was generally well tolerated, although the incidence of akathisia was greater with cariprazine than with placebo.
Trial Registration: ClinicalTrials.gov identifier: NCT01058668
J Clin Psychiatry 2015;76(3):284-292
© Copyright 2014 Physicians Postgraduate Press, Inc.
Submitted: February 21, 2014; accepted September 3, 2014.
Online ahead of print: November 25, 2014 (doi:10.4088/JCP.14m09081).
Corresponding author: Joseph R. Calabrese, MD, 10524 Euclid Ave, Room 12-135, Cleveland, OH 44106 ([email protected]).
The classic description of bipolar disorder emphasizes a cyclical nature of illness with full-blown episodes of mania and depression interspersed with periods of relative euthymia. However, more recent clinical evidence suggests that bipolar disorder may have a more chronic course characterized by mood lability, residual symptoms, sleep disturbance, cognitive impairment, and increased risk of medical and psychiatric comorbidity between major mood episodes.1
Bipolar disorder conceptualized as a chronic rather than cyclical condition necessitates an inclusive treatment paradigm defining wellness as acute remission of emergent mood episodes and amelioration of a broad range of symptoms.1 Comprehensive treatment of all aspects of bipolar disorder may result in better patient outcomes. Psychotropic medications for bipolar mania include atypical antipsychotics and mood stabilizers as first-line treatment options.2
Cariprazine, an atypical antipsychotic candidate, is an orally active and potent dopamine D3 and D2 receptor partial agonist with preferential binding to D3 receptors. The D2 receptor is known to play a fundamental role in mediating the antimanic properties of atypical antipsychotic agents.3 The D3 receptor is also thought to be involved in the modulation of mood and may present a novel target for the treatment of the broad mood symptoms associated with bipolar disorder.4-6 The high and balanced occupancy of cariprazine at both D2 and D3 receptors, which is unique to cariprazine,7 may provide distinct benefits in treating acute and mixed mania.
In a previous 3-week, phase 2 clinical trial in patients with manic or mixed episodes associated with bipolar I disorder, cariprazine 3-12 mg/d demonstrated statistically significant improvements versus placebo in improving mania symptoms and was generally well tolerated.8 The current phase 3 trial evaluated the efficacy, safety, and tolerability of low- and high-dose cariprazine in patients with acute manic or mixed episodes associated with bipolar I disorder.
METHOD
This randomized, double-blind, placebo-controlled, parallel-group, fixed/flexible-dose study in adult patients with bipolar I disorder (NCT01058668) was conducted at 65 centers in the United States, Romania, Russia, Ukraine, Croatia, and Serbia. The study was conducted from February 2010 to December 2011. Each study center received approval from appropriate ethics committees, institutional review boards, or government agencies. The study was conducted in compliance with ICH-E6 Good Clinical Practice guidelines and the Declaration of Helsinki. Participants provided written informed consent.
Patients
Male or female patients (age, 18-65 years) with bipolar I disorder, manic or mixed type, with or without psychotic symptoms based on Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR)9 criteria were eligible to enter the study. Patients had baseline Young Mania Rating Scale (YMRS)10 total score ≥ 20, score ≥ 4 on at least 2 of 4 YMRS items (irritability, speech, content, disruptive/aggressive behavior), and Montgomery-Asberg Depression Rating Scale (MADRS)11 total score < 18. Patients experiencing a first manic episode or meeting criteria for rapid cycling were excluded. Additional exclusion criteria included principal Axis I disorders other than bipolar I, cognitive/psychotic disorders, severe Axis II disorders, alcohol/substance abuse/dependence (prior 3 months), risk of suicide (ie, suicide attempt in past year, score ≥ 5 on MADRS item 10 [suicidal thoughts], significant suicide risk based on investigator judgment, or Columbia-Suicide Severity Rating Scale [C-SSRS]12 report), pregnancy/breastfeeding, or significant medical illness.
- Cariprazine 3-6 and 6-12 mg/d demonstrated significant advantage over placebo on the primary and secondary outcome measures.
- Cariprazine was generally well tolerated; the most frequent adverse event was akathisia.
- These results suggest that cariprazine, a D3 and D2 receptor partial agonist with preference for D3 receptors, may provide an effective new treatment option for patients with manic or mixed episodes associated with bipolar I disorder.
Treatment-related exclusions included electroconvulsive therapy or treatment with depot neuroleptics ≤ 3 months before study. Patients requiring treatment with prohibited medication including psychotropic drugs were excluded. Notable exceptions included lorazepam, diazepam, or oxazepam for agitation or eszopiclone, zolpidem, zolpidem extended release, chloral hydrate, or zaleplon for sleep; rescue medications for extrapyramidal symptoms (EPS) were permitted.
Study Design
The 6-week study comprised a no-drug washout period of up to 7 days, 3 weeks of double-blind treatment, and a 2-week safety follow-up. Patients were randomly assigned (1:1:1) to placebo, cariprazine 3-6 mg/d (low dose), or cariprazine 6-12 mg/d (high dose). Patients randomly assigned to cariprazine 3-6 mg/d received 1.5 mg on day 0 and 3 mg on days 1 and 2; starting on day 3, the dose could be increased in 1.5-mg increments to 6 mg/d by day 5 based on response and tolerability. Patients randomly assigned to cariprazine 6-12 mg/d received 1.5 mg on day 0, 3 mg on day 1, and 6 mg on day 2; starting on day 3, the dose could be increased in 3-mg increments to 12 mg/d by day 5. Decrease to the previous dose was allowed if there were tolerability issues. No dose increase or decrease was allowed after day 14 except for a drug holiday for up to 3 days.
All patients were hospitalized during screening and for a minimum of 2 weeks during double-blind treatment. Patients could be discharged starting on day 14 if they had a Clinical Global Impressions-Severity of Illness (CGI-S)13 score ≤ 3 (mildly ill), had no significant risk of violent/suicidal behavior, and were ready for discharge based on investigator judgment.
Efficacy Evaluations
Efficacy evaluations included the YMRS and CGI-S (screening, baseline [day 0], and days 3, 5, 7, 10, 14, and 21), CGI-Improvement (CGI-I) scale13 (days 3, 5, 7, 10, 14, and 21), MADRS (screening and days 0, 7, 14, and 21), and Positive and Negative Syndrome Scale (PANSS)14 (days 0, 7, 14, and 21). Safety was assessed via treatment-emergent adverse events (TEAEs), clinical laboratory evaluations, vital signs, electrocardiograms, EPS scales (Barnes Akathisia Rating Scale [BARS],15 Simpson-Angus Scale [SAS],16 and Abnormal Involuntary Movement Scale [AIMS]),17 and C-SSRS.
Statistical Analyses
All efficacy analyses were based on the ITT population, defined as all patients who received study drug and had ≥ 1 postbaseline assessment of the YMRS total score.
The primary and secondary efficacy parameters were mean change from baseline to week 3 in YMRS total score and CGI-S score, respectively. Comparison of cariprazine 3-6 mg/d and 6-12 mg/d versus placebo was performed using a mixed-effects model for repeated measures (MMRM) with treatment group, study center, visit, and treatment group-by-visit interaction as fixed effects and the baseline value and baseline-by-visit interaction as the covariates; an unstructured covariance matrix was used to model the covariance of within-patient scores. Sensitivity analyses for the primary efficacy parameter used a pattern-mixture model (PMM) based on nonfuture-dependent missing value restrictions18 and analysis of covariance model with treatment group and study center as factors and baseline YMRS total score as the covariate with missing values imputed using the last-observation-carried-forward (LOCF) approach. YMRS effect sizes (Cohen d) were calculated post hoc for MMRM and LOCF models. Additionally, post hoc analysis was conducted to evaluate change from baseline to week 3 for all YMRS single items using an MMRM model.
Additional efficacy parameters (MADRS and PANSS total score change from baseline to week 3, CGI-I score at week 3) were analyzed using an MMRM model similar to the primary and secondary analyses. YMRS response (≥ 50% total score improvement) and remission (total score ≤ 12) rates at week 3 were analyzed using a logistic regression model with treatment group and corresponding baseline score as explanatory variables with missing values imputed using the LOCF approach; number needed to treat (NNT) estimates for YMRS response and remission were calculated post hoc. Post hoc analyses evaluated remission rates using a more stringent criterion (YMRS total score < 8).
Tests for statistical significance were performed at the 2-sided 5% significance level; confidence intervals (CIs) were 2-sided 95% CIs. To control for multiple comparisons for the primary and secondary efficacy parameters, a matched parallel gatekeeping procedure19 was used. Significance of the secondary endpoint for each dose would not be claimed unless the corresponding primary outcome was significant. Additional and by-visit efficacy analyses were not controlled for multiple comparisons. Comparisons between cariprazine groups and placebo for mean change from baseline in the individual YMRS items were controlled for multiple comparisons using the Hochberg procedure.20 For all efficacy parameters, statistical significance was defined as P values < .05.
All safety measures were based on the safety population, defined as all patients who received at least 1 dose of study medication. TEAEs were analyzed using descriptive statistics (n [%]). Post hoc statistical testing was performed for mean changes from baseline in clinical laboratory parameters, vital signs, and extrapyramidal symptom scales; P values were based on a 2-sample t test. Treatment-emergent EPS (parkinsonism) was defined as SAS score ≤ 3 at baseline and > 3 at any postbaseline visit; treatment-emergent akathisia was defined as BARS score ≤ 2 at baseline and > 2 at any postbaseline visit; P values were based on χ2 testing.
RESULTS
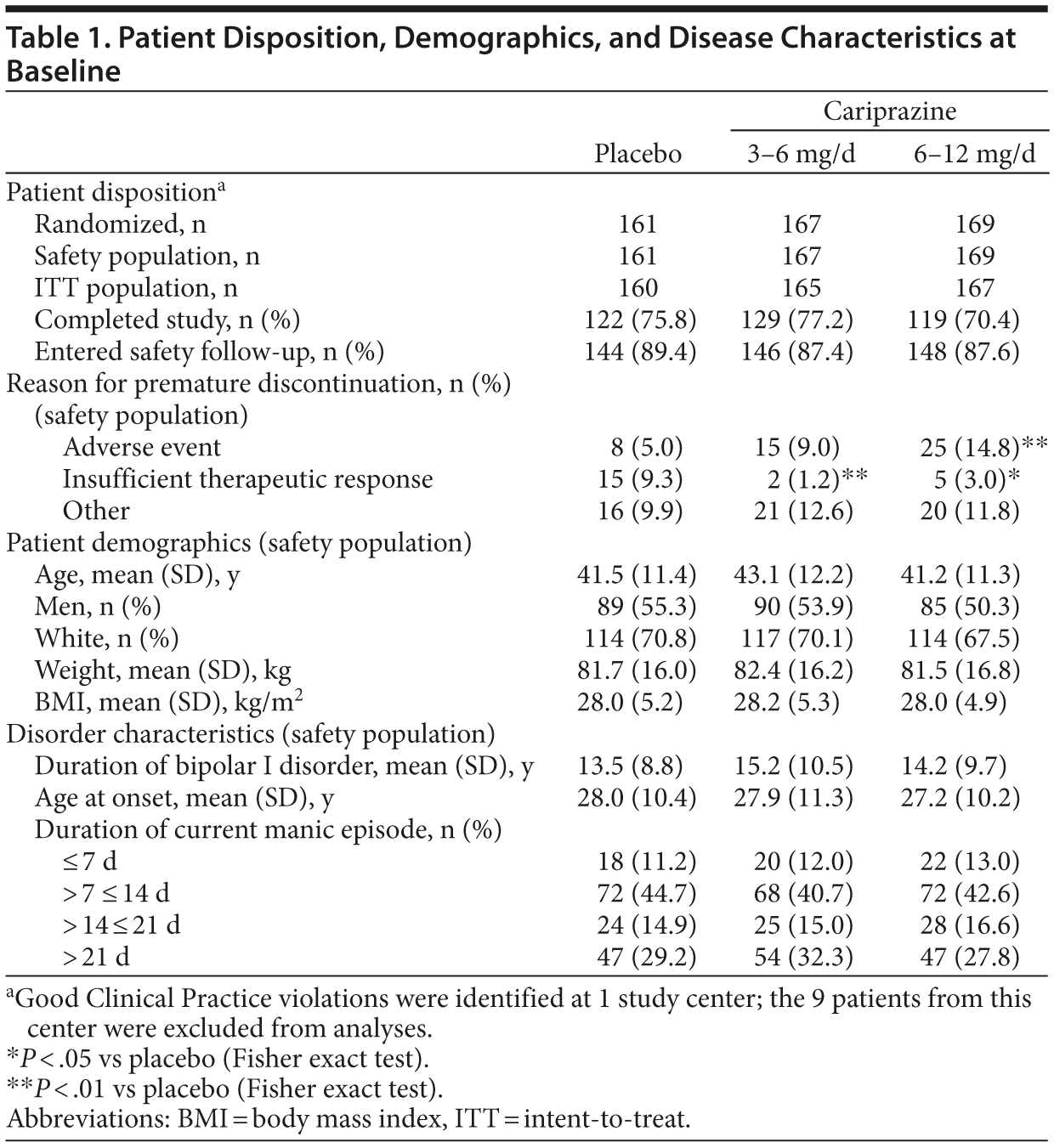
The majority of patients were enrolled at study centers in the United States (54%). Patient disposition and demographics are summarized in Table 1. Rates of premature discontinuation were similar between groups. Significantly more cariprazine 6-12 mg/d patients than placebo patients discontinued due to adverse events. Significantly more placebo patients than cariprazine patients discontinued due to insufficient therapeutic response.
Click figure to enlarge
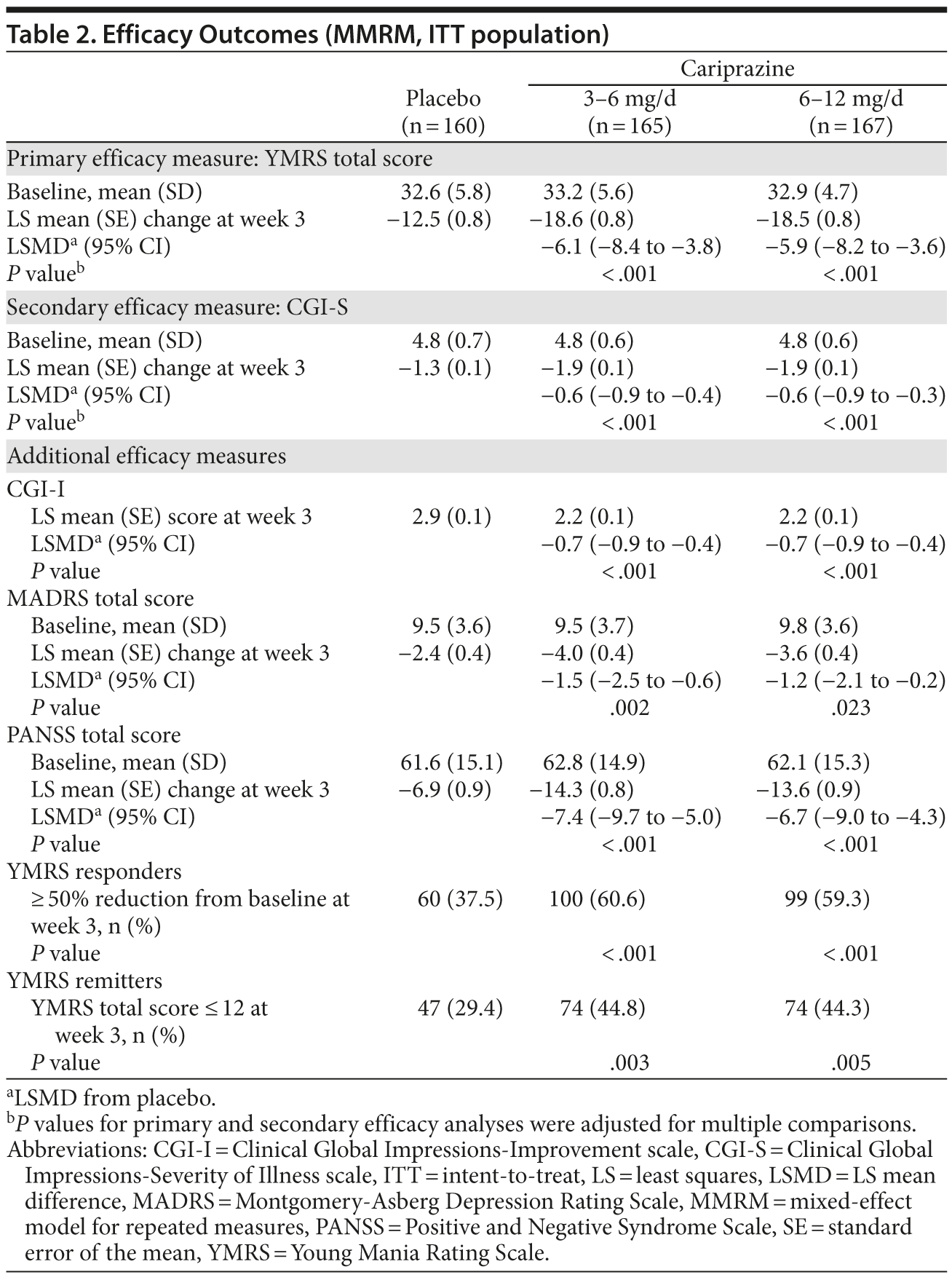
Baseline characteristics and bipolar history were similar among groups (Table 1). Mean YMRS, CGI-S, and PANSS scores at baseline indicated moderate to severe illness10,13,21; MADRS scores indicated low levels of depression at baseline (Table 2).
Click figure to enlarge
Efficacy
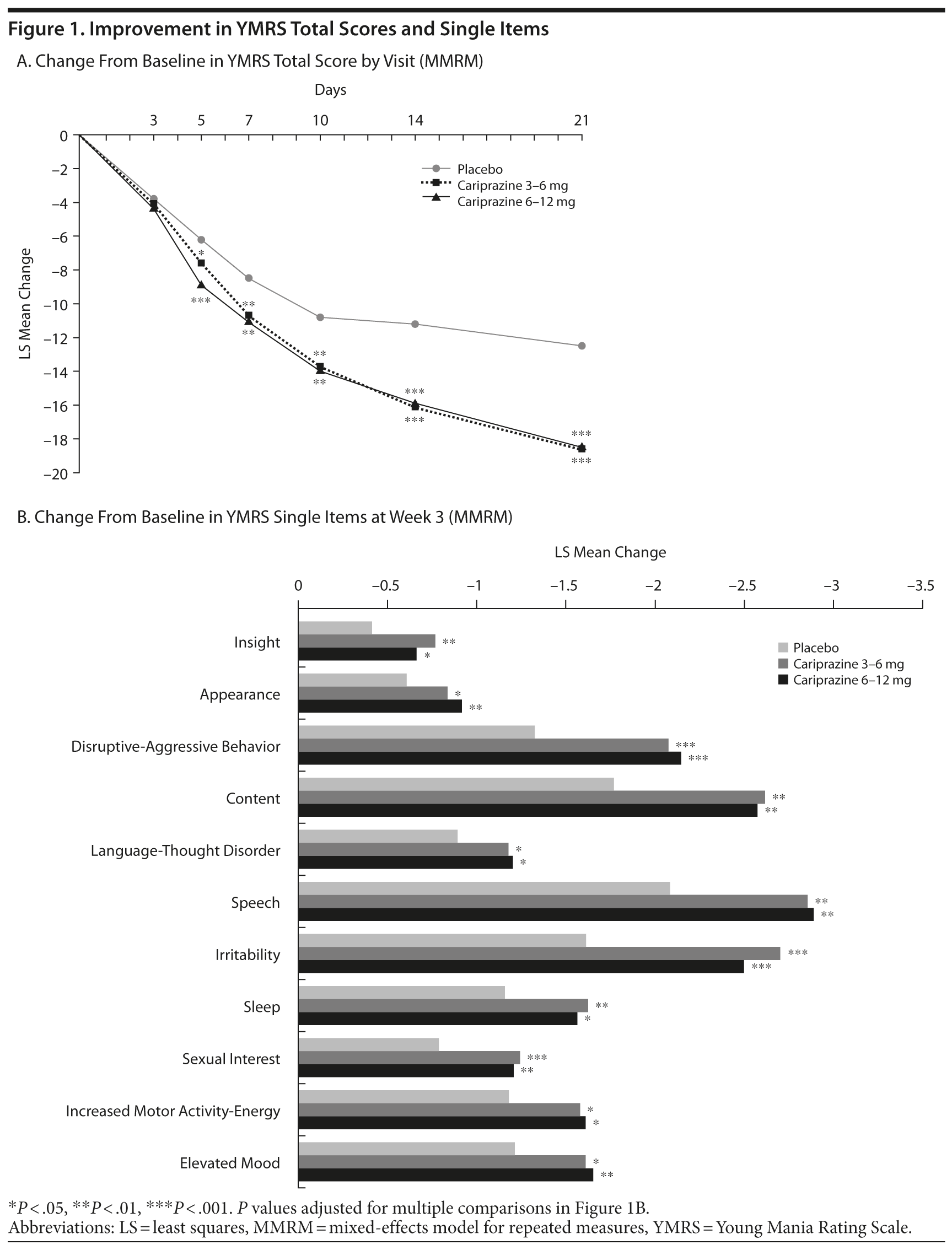
YMRS total score change from baseline to week 3 was statistically significantly greater for both cariprazine groups compared with placebo (Figure 1A, Table 2). Primary results were supported by PMM and LOCF sensitivity analyses (P < .001 for both cariprazine groups vs placebo for all PMM location shifts and week 3 LOCF; data not shown). Statistically significant separation from placebo on YMRS total score was observed at every visit from day 5 through day 21 (Figure 1A) for both cariprazine groups. CGI-S total score mean change from baseline was statistically significantly greater for both cariprazine groups versus placebo (Table 2).
Click figure to enlarge
Post hoc analysis of primary results yielded effect sizes of 0.62 and 0.60 for cariprazine 3-6 mg/d and 6-12 mg/d, respectively, using the MMRM approach. LOCF effect sizes were 0.58 and 0.53 for the cariprazine 3-6 and 6-12 mg/d groups, respectively. Post hoc analyses of YMRS single items showed statistically significant superiority of both cariprazine groups versus placebo following multiplicity adjustment on all 11 items (Figure 1B).
Statistically significant differences between both cariprazine doses versus placebo were also seen on all other efficacy parameters at week 3 (Table 2). A greater percentage of patients in the low- and high-dose cariprazine groups compared with placebo met YMRS response and remission criteria at week 3 (Table 2). For the cariprazine 3-6 mg/d group, the NNT estimates for response and remission were 5 (95% CI, 3 to 8) and 7 (95% CI, 4 to 20), respectively. Similar results were observed in the cariprazine 6-12 mg/d group, with NNT estimates of 5 (95% CI, 4 to 9) for response and 7 (95% CI, 4 to 22) for remission. Using the more stringent cutoff for remission (YMRS score < 8), a significantly greater percentage of patients achieved remission in the 6-12 mg/d group (25%) than placebo (16%) at week 3 (P = .039); remission rates in the cariprazine 3-6 mg/d group (22%) were also greater than placebo (16%), but the difference did not achieve statistical significance.
Safety
The final mean daily doses for the cariprazine 3-6 mg/d and 6-12 mg/d groups were 4.8 mg and 9.1 mg, respectively. In the cariprazine 3-6 mg/d and 6-12 mg/d groups, 74% of patients received 6 mg/d and 70% of patients received 12 mg/d at the final visit, respectively.
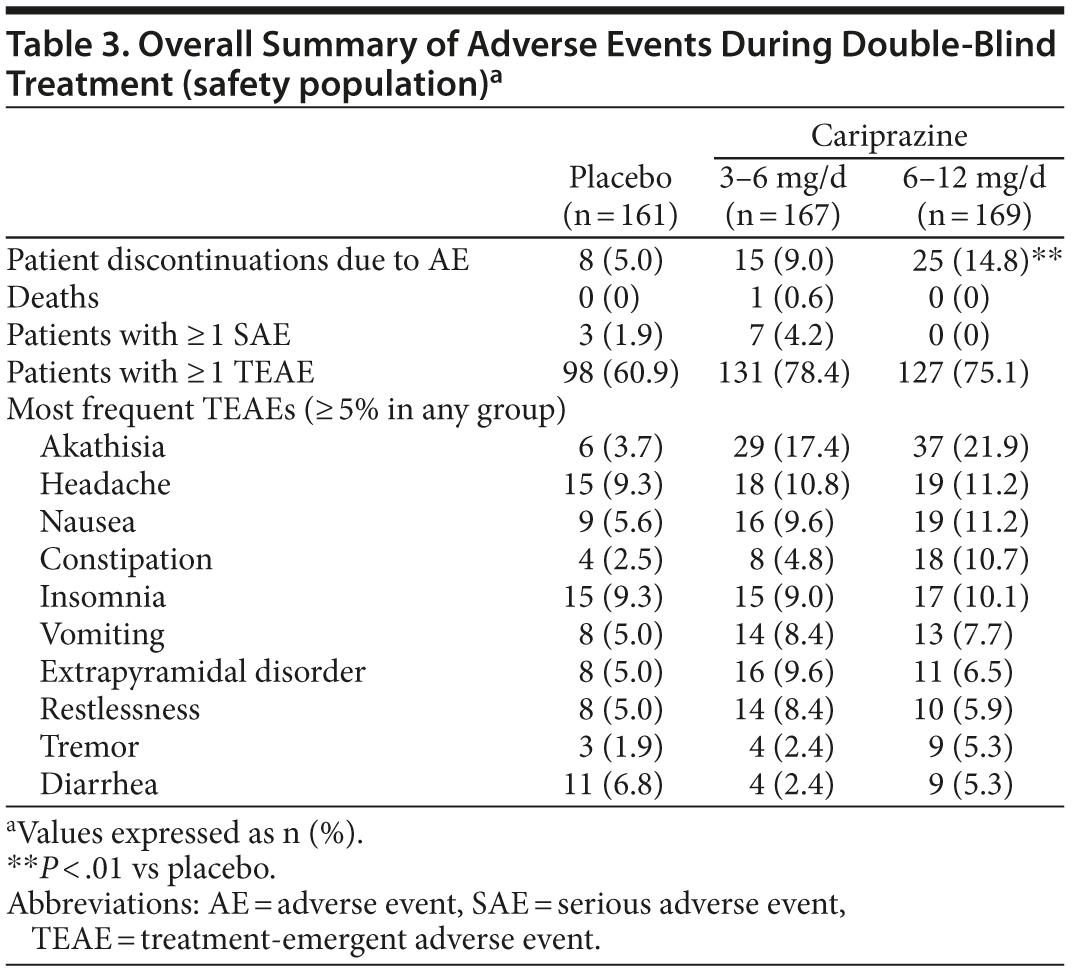
Adverse events. An overall summary of AEs is presented in Table 3. Common cariprazine TEAEs (≥ 5% in either cariprazine group and twice placebo) were akathisia (both cariprazine doses) and nausea, constipation, and tremor (cariprazine 6-12 mg/d only). The majority of TEAEs were considered by the investigator to be mild or moderate in intensity.
Click figure to enlarge
The most common AEs leading to discontinuation were mania (3 [2%] placebo, 3 [2%] cariprazine 3-6 mg/d, and 2 [1%] cariprazine 6-12 mg/d patients) and akathisia (0 placebo, 3 [2%] cariprazine 3-6 mg/d, and 5 [3%] cariprazine 6-12 mg/d patients). All 3 serious adverse events in the placebo group (mania, bipolar disorder, and bipolar I disorder) and 4 of 7 in the cariprazine 3-6 mg/d group (mania [2], bipolar disorder, aggression) were associated with worsening of mania or bipolar disorder. There were no SAEs in the cariprazine 6-12 mg/d group. SAEs led to premature discontinuation in 0 placebo patients and 4 cariprazine 3-6 mg/d patients (2%; mania [2 patients], aggression, and bipolar disorder). One death from pulmonary embolism occurred in a female patient with a history of mild hypertension; she received double-blind cariprazine 3-6 mg/d for 8 days before study discontinuation due to insufficient therapeutic response. The death occurred 9 days after discontinuation of study drug and was not considered related to treatment.
Benzodiazepine use to control agitation was similar between cariprazine groups (3-6 mg/d, 64%; 6-12 mg/d, 63%) and placebo (58%).
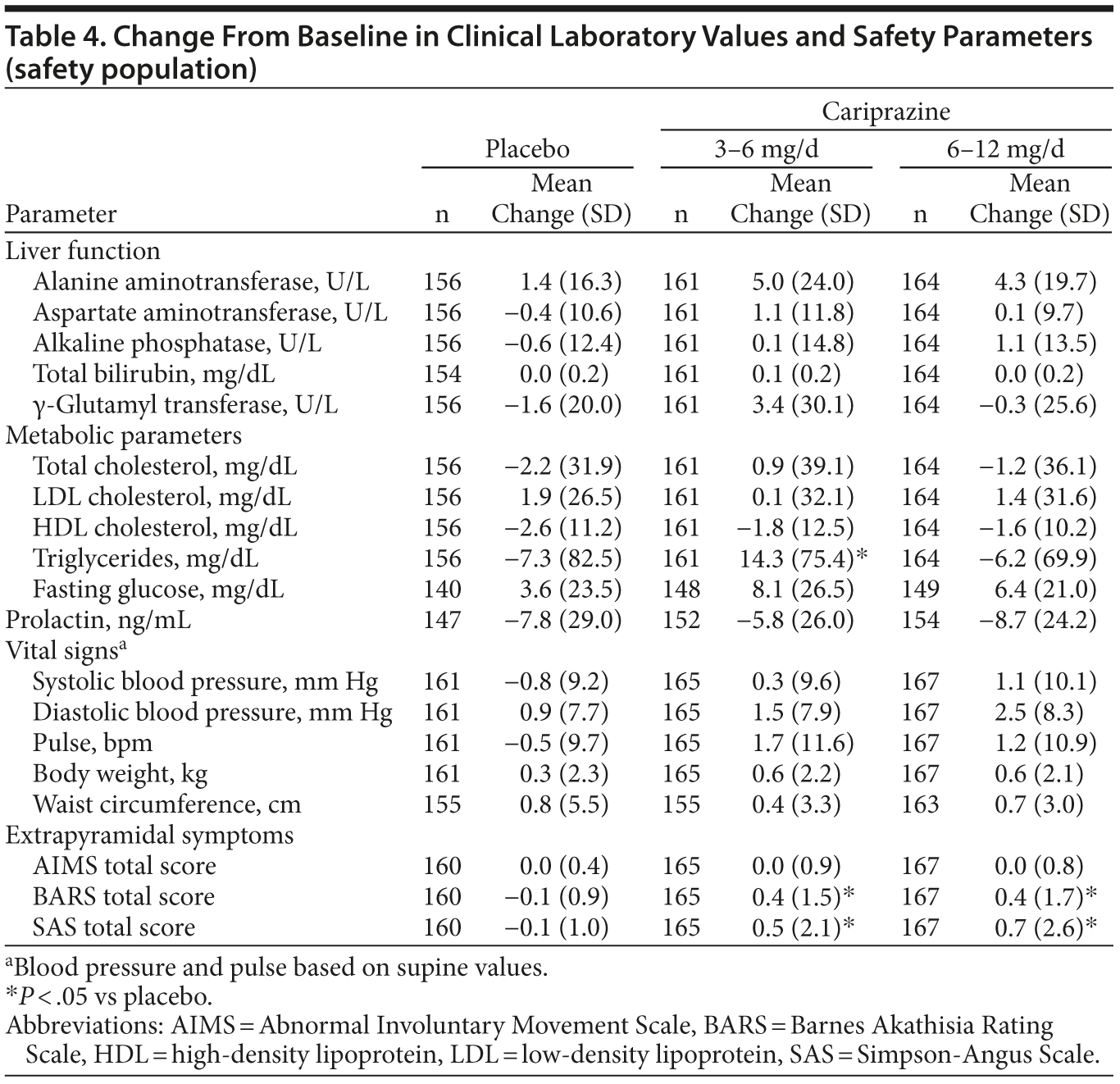
Laboratory parameters. There were no statistically significant differences between cariprazine and placebo for mean change from baseline in metabolic parameters, liver enzymes, or prolactin (Table 4), with the exception of increased triglycerides in the 3-6 mg/d group (mean [SD] change: 3-6 mg/d, 14.3 mg/dL [75.4]; placebo, −7.3 mg/dL [82.5]; P = .015); mean (SD) change in triglyceride levels in the cariprazine 6-12 mg/d (−6.2 mg/dL [69.9]) was similar to placebo (P = .889). No patient met Hy’s Law criteria (ALT or aspartate aminotransferase [AST] ≥ 3 ×— upper limit of normal [ULN], total bilirubin ≥ 2 ×— ULN, and alkaline phosphatase < 2 ×— ULN).22
Click figure to enlarge
Vital signs and cardiac safety. Mean changes in vital signs were similar across groups (Table 4). No cariprazine-treated patient had a potentially clinically significant increase in systolic blood pressure (≥ 180 mm Hg and increase ≥ 20 mm Hg) or pulse rate (≥ 120 bpm and increase ≥ 15 bpm). Potentially clinically significant increases in diastolic blood pressure (≥ 105 mm Hg and increase ≥ 15 mm Hg) occurred in only 1 placebo patient (0.6%) and 1 cariprazine 3-6 mg/d patient (0.6%). The percentage of potentially clinically significant changes in body weight (> 7% increase or decrease) was generally small and similar between groups (placebo, 2%; 3-6 mg/d, 1%; 6-12 mg/d, 2%). The incidence of orthostatic hypotension was similar between groups (placebo, 12%; 3-6 mg/d, 12%; 6-12 mg/d, 9%). No patient in any treatment group had a Fridericia QTc interval of > 500 msec; 1 patient each in the placebo and cariprazine 6-12 mg/d groups had a Bazett QTc interval of > 500 msec.
Extrapyramidal symptoms. Mean changes in AIMS total score were similar between groups; mean change in BARS and SAS total scores was significantly higher in both cariprazine groups relative to placebo (Table 4). Significantly more patients in the cariprazine groups relative to placebo met criteria for treatment-emergent EPS (parkinsonism) (placebo, 1%; 3-6 mg/d, 11% [P < .001]; 6-12 mg/d, 14% [P < .001]) and akathisia (placebo, 4%; 3-6 mg/d, 20% [P < .001]; 6-12 mg/d, 23% [P < .001]).
Discontinuations due to EPS-related AEs including akathisia/restlessness occurred in 11 patients (3 [2%] cariprazine 3-6 mg/d and 8 [5%] cariprazine 6-12 mg/d); none of the AEs were classified as SAEs. Most EPS-related TEAEs were mild or moderate in intensity (placebo, 96%; 3-6 mg/d, 93%; 6-12 mg/d, 93%). Use of antiparkinson medication was higher in the cariprazine treatment groups (both groups, 17%) than in placebo (7%); use of β-blocking agents was also higher in the cariprazine groups (3-6 mg/d, 4%; 6-12 mg/d, 5%) versus the placebo group (1%).
Suicidal ideation and behavior. Based on C-SSRS assessments, no suicidal behavior was noted; suicidal ideation was recorded for 1.9% of placebo-treated patients and 1.2% and 2.4% of cariprazine 3-6 mg/d and 6-12 mg/d patients, respectively. Suicidal ideation AEs were reported in 1 placebo patient and 0 cariprazine patients.
DISCUSSION
In this phase 3 study, both low (3-6 mg/d) and high (6-12 mg/d) doses of cariprazine demonstrated efficacy and were generally well tolerated in adult patients with acute manic or mixed mania associated with bipolar disorder. Both cariprazine groups showed significant superiority over placebo on the primary efficacy parameter, mean change from baseline to week 3 on YMRS scores. Statistical superiority occurred early and was maintained through the end of treatment in both cariprazine groups.
Improvement on the CGI-S, CGI-I, PANSS, and MADRS was also statistically greater for both cariprazine doses versus placebo. These results suggest that cariprazine-treated patients improved in global disease severity and did not show worsening or exacerbation of depressive or psychotic symptoms.
Treatment effect sizes for YMRS improvement were similar for the low-dose (0.62, MMRM; 0.58, LOCF) and high-dose (0.60, MMRM; 0.53, LOCF) cariprazine groups. There was no active comparator in this trial, so no direct comparisons to other antipsychotics could be made. Indirect comparison was possible, based on a meta-analysis of 29 trials (N = 7,295) in mania. This analysis found an overall effect size of 0.40 (95% CI, 0.32 to 0.47; P < .0001) for atypical antipsychotics as a group versus placebo.23 Cariprazine data from the previous phase 2 study in acute or mixed mania8 were included in this meta-analysis; the largest effect sizes for atypical antipsychotics versus placebo in this study were risperidone and cariprazine (effect size: 0.66 [3 trials] and 0.51 [1 trial], respectively), with at least moderate effect sizes observed for the other atypical agents (range, 0.26-0.46). The current phase 3 results support the efficacy of cariprazine seen in the meta-analyses.
Treatment response as opposed to disease remission is highly correlated with the occurrence of residual symptoms, rapid relapse, and more chronic illness,24,25 and therefore remission is considered the treatment goal in bipolar mania. Although no standardized definition for remission exists, a virtual lack of symptoms is most commonly operationalized as a YMRS cutoff score ≤ 12.24 Using this definition of remission, statistically significantly greater percentages of cariprazine patients in both the low- and high-dose groups (45% and 44%) versus placebo (29%) achieved remission at week 3; the NNT was 7 for both groups. Cariprazine compared favorably to pooled data that reported remission rates of 40%-50% for risperidone,26 quetiapine,27 and olanzapine28; a recent meta-analysis determined that the NNT for remission for atypical antipsychotics as a group was 8.29 While a YMRS cutoff of ≤ 12 is the most commonly used definition of remission in clinical trials, more stringent definitions may better identify patients that are truly asymptomatic.24 In a recent publication, the International Society for Bipolar Disorders recommended a YMRS score < 8 for measuring remission in bipolar mania, as this cutoff may be more representative of minimal symptomatology and a patient’s ability to function.30 Using the more stringent YMRS score < 8 criterion, greater rates of remission were observed in the cariprazine 6-12 mg/d group (25%) compared with placebo (16%).
Post hoc investigation of YMRS single items revealed significant improvements in both low- and high-dose cariprazine groups versus placebo on all 11 YMRS items. Statistical significance versus placebo was maintained on all YMRS items for both cariprazine dose groups following adjustment for multiple comparisons. These results confirmed findings from a previous phase 2 cariprazine study, which showed significant effects with cariprazine 3-12 mg/d versus placebo across the entire spectrum of YMRS-measured mania symptoms.8 As unresolved symptoms are common following antipsychotic treatment, the remission data and broad efficacy seen across mania symptoms following cariprazine treatment are particularly encouraging.
Cariprazine was generally well tolerated. Discontinuations due to adverse events and incidences of akathisia were more frequent in the cariprazine 6-12 mg/d group relative to the 3-6 mg/d group. The tolerability profile was generally similar between dose groups on other safety parameters. The most common AEs leading to discontinuation were mania and akathisia. The incidence of SAEs was low in all 3 groups (2% placebo, 4% low-dose cariprazine, and 0% high-dose cariprazine); most SAEs were related to the worsening of mania or bipolar disorder.
Similar to some atypical antipsychotics, cariprazine was associated with higher incidence of EPS relative to placebo. The only EPS-related TEAEs occurring at an incidence of ≥ 5% and twice the rate of placebo were akathisia and tremor. Akathisia was reported in approximately 17% and 22% of patients in the cariprazine 3-6 and 6-12 mg/d groups, respectively, compared with 4% of patients in the placebo group. Akathisia resulted in premature discontinuation of treatment in approximately 2% and 3% of patients in the cariprazine 3-6 and 6-12 mg/d groups, respectively. No other EPS-related AE resulted in discontinuation of ≥ 2% of patients in any treatment group. Incidences of treatment-emergent EPS (parkinsonism) and akathisia per the SAS and BARS, respectively, were also more common in the cariprazine groups versus the placebo group. EPS-related TEAEs were generally classified as mild or moderate (approximately 93% in each cariprazine group) in intensity.
Cardiovascular disease is responsible for the largest total number of excess deaths in bipolar disorder, with risk factors almost twice as prevalent in bipolar patients versus the general population.31 The risk for cardiovascular disease in bipolar disorder can exist independently of the treatment used to manage it, although medications may exacerbate some risks.32 In this study, mean changes from baseline in metabolic parameters (eg, cholesterol, triglycerides, fasting glucose) were similar among groups, with the exception of increased triglyceride levels in the cariprazine 3-6 mg/d group relative to the placebo group. Mean changes in body weight and waist circumference were small and similar for cariprazine and placebo; however, as the study duration was only 3 weeks, these changes should be interpreted accordingly. Mean changes in vital signs were also generally similar among groups; no QTc interval over 500 msec was observed in any treatment group.
This study was limited by the lack of an active comparator, and the short study duration limits analyses of longer-term outcomes. Additionally, conclusions regarding the risk/benefit profile of the different cariprazine doses are difficult due to the flexible-dose design and the lack of power to detect differences between cariprazine dose groups. However, clinicians should take into account individual patient differences and response and tolerability to medication when selecting the appropriate cariprazine dosage for treatment.
CONCLUSION
In patients with manic or mixed episodes associated with bipolar I disorder, cariprazine showed statistically significant improvement versus placebo on the primary, secondary, and all additional efficacy parameters.
Cariprazine was generally well tolerated and exhibited a favorable metabolic profile; incidences of akathisia were greater with cariprazine treatment than with placebo. These results support findings from a previous study8 in acute and mixed mania suggesting that cariprazine, a D3 receptor-preferring D3 and D2 partial agonist antipsychotic candidate, may be a valuable new treatment option for bipolar I disorder.
Drug names: diazepam (Diastat, Valium, and others), eszopiclone (Lunesta), lorazepam (Ativan and others), olanzapine (Zyprexa), quetiapine (Seroquel), risperidone (Risperdal and others), zaleplon (Sonata and others), zolpidem (Ambien, Edluar, and others).
Author affiliations: University Hospitals Case Medical Center, Case Western Reserve School of Medicine, Cleveland, Ohio (Dr Calabrese); Lindner Center of HOPE, Mason, and Department of Psychiatry & Behavioral Neuroscience, University of Cincinnati College of Medicine, Cincinnati, Ohio (Dr Keck); Forest Research Institute, Jersey City, New Jersey (Ms Starace and Drs Lu and Durgam); Prescott Medical Communications Group, Chicago, Illinois (Dr Ruth); and Gedeon Richter Plc, Budapest, Hungary (Drs Laszlovszky and Németh).
Potential conflicts of interest: Dr Calabrese has received federal funding from the Department of Defense, Health Resources Services Administration, and National Institute of Mental Health; received research support from Abbott, AstraZeneca, Bristol-Myers Squibb, Cephalon, Cleveland Foundation, Eli Lilly, GlaxoSmithKline, Janssen, NARSAD, Repligen, Stanley Medical Research Institute, Takeda, and Wyeth; served on advisory boards of Abbott, AstraZeneca, Bristol-Myers Squibb, Cephalon, Dainippon Sumitomo, EPI-Q, Forest, France Foundation, Gedeon Richter, GlaxoSmithKline, Janssen, Johnson and Johnson, Lundbeck, Merck, Neurosearch, Ortho-McNeil, Otsuka, Pfizer, Repligen, Schering-Plough, Servier, Solvay, Supernus, Synosia, Takeda, and Wyeth; and provided CME lectures supported by AstraZeneca, Bristol-Myers Squibb, France Foundation, GlaxoSmithKline, Janssen, Johnson and Johnson, Merck, Sanofi Aventis, Schering-Plough, Pfizer, Solvay, and Wyeth. Dr Keck has received consulting or advisory board fees from Forest, Shire, Sunovion, and Alkermes. Ms Starace and Drs Lu and Durgam are employees of Forest Research Institute. Dr Ruth is an employee of Prescott Medical Communications Group, a contractor for Forest Research Institute. Drs Laszlovszky and Németh are employees of Gedeon Richter.
Funding/support: Supported by funding from Forest Laboratories, Inc (New York, New York) and Gedeon Richter Plc (Budapest, Hungary).
Role of the sponsors: Forest Laboratories, Inc and Gedeon Richter Plc were involved in the study design; collection (via contracted clinical investigator sites), analysis, and interpretation of data; and the decision to present these results.
Previous presentations: American Psychiatric Association, May 18-22, 2013; San Francisco, California; New Clinical Drug Evaluation Unit, May 28-31, 2013; Hollywood, Florida; International Conference on Bipolar Disorders; June 13-16, 2013; Miami Beach, Florida; US Psychiatric and Mental Health Congress; September 30-October 3, 2013; Las Vegas, Nevada; and European College of Neuropsychopharmacology, October 5-9, 2013; Barcelona, Spain.
Acknowledgments: The authors acknowledge the investigators at each study center: United States: Ronald L. Brenner, MD, Neurobehavioral Research Inc, Cedarhurst, NY; Barbara A. Burtner, MD, Accurate Clinical Trials, Inc, Kissimmee, FL; Daniel Chueh, MD, Neuropsychiatric Research Center of Orange County, Santa Ana, CA; David Feifel, MD, PhD, UCSD Medical Center-Hillcrest, San Diego, CA; Robert E. Litman, MD, CBH Health, LLC, Rockville, MD; Mark A. Novitsky, MD, CRI Worldwide, LLC, Philadelphia, PA; Robert A. Riesenberg, MD, Atlanta Center for Medical Research, Atlanta, GA; David Struble, MD, Research Strategies of Memphis, LLC, Memphis, TN. Romania: George Badescu, MD, Spitalul Clinic de Neuropsihiatrie Craiova, Craiova, Eda Ciorabai, MD, Spitalul Clinic Judetean de Urgenta Constanta, Constanta; Maria Ladea, MD, Spitalul Clinic de Psihiatrie “Prof Dr Alexandru Obregia, Bucuresti”; Delia Podea, MD, PhD, Spitalul Clinic Judetean de Urgenta Arad, Arad. Ukraine: Yuliya Blazhevych, MD, Kyiv City Clinical Psychoneurological Hospital #1, Kyiv; Svitlana Kazakova, MD, Lugansk Regional Clinical Psychoneurological Hospital, Lugansk State Medical University, Lugansk; Svitlana Moroz, MD, Ukrainian State Scientific and Research Institute of Medical and Social Problems of Disability, Dnipropetrovsk; Katerina Zakal, MD, Municipal Institution “Lviv State Regional Clinical Psychiatric Hospital” Lviv. Serbia: Miroslava Jasovic-Gasic, MD, PhD, Institute for Psychiatry, Clinical Center of Serbia, Belgrade; Dusica Lecic’ Tosevski, MD, PhD, Institute of Mental Health, Belgrade. In Russia: Vladimir Sheshenin, PhD, Mental Health Research of Russian Academy of Medical Sciences, Moscow. Croatia: Gordan Makaric, MD, University Department of Psychiatry, Psychiatric Hospital VrapÄe, Zagreb; Gordana Rubesa, MD, PhD, University Hospital Center Rijeka, Rijeka. Writing assistance and editorial support for the preparation of this manuscript were provided by Carol Dyer, MS, and Paul Ferguson, MS, of Prescott Medical Communications Group, Chicago, Illinois, contractors of Forest Research Institute.
REFERENCES
1. Leboyer M, Kupfer DJ. Bipolar disorder: new perspectives in health care and prevention. J Clin Psychiatry. 2010;71(12):1689-1695. PubMed doi:10.4088/JCP.10m06347yel
2. Yatham LN, Kennedy SH, Parikh SV, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2013. Bipolar Disord. 2013;15(1):1-44. PubMed doi:10.1111/bdi.12025
3. Harrison-Read PE. Antimanic potency of typical neuroleptic drugs and affinity for dopamine D2 and serotonin 5-HT2A receptors—a new analysis of data from the archives and implications for improved antimanic treatments. J Psychopharmacol. 2009;23(8):899-907. PubMed doi:10.1177/0269881108094349
4. Beaulieu JM, Tirotta E, Sotnikova TD, et al. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J Neurosci. 2007;27(4):881-885. doi:10.1523/JNEUROSCI.5074-06.2007 PubMed
5. Cho DI, Zheng M, Kim KM. Current perspectives on the selective regulation of dopamine D₂ and D₃ receptors. Arch Pharm Res. 2010;33(10):1521-1538. PubMed doi:10.1007/s12272-010-1005-8
6. Leggio GM, Salomone S, Bucolo C, et al. Dopamine D(3) receptor as a new pharmacological target for the treatment of depression. Eur J Pharmacol. 2013;719(1-3):25-33. PubMed doi:10.1016/j.ejphar.2013.07.022
7. Kiss B, Horti F, Bobok A. Cariprazine, a D3/D2 dopamine receptor partial agonist antipsychotic, displays greater D3 receptor occupancy in vivo compared with other antipsychotics. Schizophr Res. 2012;136(supplement 1):S190. doi:10.1016/S0920-9964(12)70588-1
8. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial [published online ahead of print July 24, 2014]. Bipolar Disord. PubMed doi:10.1111/bdi.12238
9. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
10. Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133(5):429-435. PubMed doi:10.1192/bjp.133.5.429
11. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382-389. PubMed doi:10.1192/bjp.134.4.382
12. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266-1277. PubMed doi:10.1176/appi.ajp.2011.10111704
13. Guy W. The Clinical Global Impression Severity and Improvement Scales. ECDEU Assessment Manual for Psychopharmacology. US Department of Health, Education and Welfare publication (ADM). 76-338. Rockville, MD: National Institute of Mental Health; 1976:218-222.
14. Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261-276. PubMed doi:10.1093/schbul/13.2.261
15. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672-676. PubMed doi:10.1192/bjp.154.5.672
16. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand suppl. 1970;45(S212):11-19. PubMed doi:10.1111/j.1600-0447.1970.tb02066.x
17. Guy W. The Abnormal Involuntary Movement Scale. ECDEU Assessment Manual for Psychopharmacology. US Department of Health, Education and Welfare publication (ADM). 76-338. Rockville, MD: National Institute of Mental Health; 1976:218-222.
18. Kenward MG, Molenberghs G, Thijs H. Pattern-mixture models with proper time dependence. Biometrika. 2003;90(1):53-71. doi:10.1093/biomet/90.1.53
19. Chen X, Luo X, Capizzi T. The application of enhanced parallel gatekeeping strategies. Stat Med. 2005;24(9):1385-1397. PubMed doi:10.1002/sim.2005
20. Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9(7):811-818. PubMed doi:10.1002/sim.4780090710
21. Leucht S, Kane JM, Kissling W, et al. What does the PANSS mean? Schizophr Res. 2005;79(2-3):231-238. PubMed doi:10.1016/j.schres.2005.04.008
22. Watkins PB, Seligman PJ, Pears JS, et al. Using controlled clinical trials to learn more about acute drug-induced liver injury. Hepatology. 2008;48(5):1680-1689. PubMed doi:10.1002/hep.22633
23. Yildiz A, Vieta E, Leucht S, et al. Efficacy of antimanic treatments: meta-analysis of randomized, controlled trials. Neuropsychopharmacology. 2011;36(2):375-389. PubMed doi:10.1038/npp.2010.192
24. Beyer JL. An evidence-based medicine strategy for achieving remission in bipolar disorder. J Clin Psychiatry. 2008;69(suppl 3):31-37. PubMed
25. Judd LL, Schettler PJ, Akiskal HS, et al. Residual symptom recovery from major affective episodes in bipolar disorders and rapid episode relapse/recurrence. Arch Gen Psychiatry. 2008;65(4):386-394. PubMed doi:10.1001/archpsyc.65.4.386
26. Gopal S, Steffens DC, Kramer ML, et al. Symptomatic remission in patients with bipolar mania: results from a double-blind, placebo-controlled trial of risperidone monotherapy. J Clin Psychiatry. 2005;66(8):1016-1020. PubMed doi:10.4088/JCP.v66n0809
27. Vieta E, Mullen J, Brecher M, et al. Quetiapine monotherapy for mania associated with bipolar disorder: combined analysis of two international, double-blind, randomised, placebo-controlled studies. Curr Med Res Opin. 2005;21(6):923-934. PubMed doi:10.1185/030079905X46340
28. Ketter TA, Wang P, Nowakowska C, et al. Advances in the treatment of acute mania. In: Ketter TA, Calabrese JR, eds. Advances in Treatment of Bipolar Disorders: Review of Psychiatry Series. Washington, DC: American Psychiatric Association; 2005:11-56.
29. Tamayo JM, Zarate CA Jr, Vieta E, et al. Level of response and safety of pharmacological monotherapy in the treatment of acute bipolar I disorder phases: a systematic review and meta-analysis. Int J Neuropsychopharmacol. 2010;13(6):813-832. PubMed doi:10.1017/S1461145709991246
30. Tohen M, Frank E, Bowden CL, et al. The International Society for Bipolar Disorders (ISBD) Task Force report on the nomenclature of course and outcome in bipolar disorders. Bipolar Disord. 2009;11(5):453-473. PubMed doi:10.1111/j.1399-5618.2009.00726.x
31. Birkenaes AB, Opjordsmoen S, Brunborg C, et al. The level of cardiovascular risk factors in bipolar disorder equals that of schizophrenia: a comparative study. J Clin Psychiatry. 2007;68(6):917-923. PubMed doi:10.4088/JCP.v68n0614
32. Maina G, Salvi V, Vitalucci A, et al. Prevalence and correlates of overweight in drug-naׯve patients with bipolar disorder. J Affect Disord. 2008;110(1-2):149-155. PubMed doi:10.1016/j.jad.2007.12.233
Download Free PDF
This PDF is free for all visitors!