Objective: To test the efficacy of current treatment recommendations for parkinsonism and tardive dyskinesia (TD) severity in patients with severe mental illness (SMI).
Methods: We present an 18-year prospective study including all 223 patients with SMI (as defined by the 1987 US National Institute of Mental Health, which were based on DSM-III-R diagnostic criteria) receiving care from the only psychiatric hospital of the former Netherlands Antilles. Eight clinical assessments (1992-2009) focused on movement disorders and medication use. Tardive dyskinesia was measured by the Abnormal Involuntary Movement Scale and parkinsonism by the Unified Parkinson’s Disease Rating Scale. Antipsychotics were classified into first-generation antipsychotic (FGA) versus second-generation antipsychotic (SGA) and high versus low dopamine 2 (D2) affinity categories. The effect that switching has within each category on subsequent movement scores was calculated separately by using time-lagged multilevel logistic regression models.
Results: There was a significant association between reduction in TD severity and starting/switching to an FGA (B = −3.54, P < .001) and starting/switching to a high D2 affinity antipsychotic (B = −2.49, P < .01). Adding an SGA to existing FGA treatment was associated with reduction in TD severity (B = −2.43, P < .01). For parkinsonism, stopping antipsychotics predicted symptom reduction (B = −7.76, P < .01 in FGA/SGA-switch model; B = −7.74, P < .01 in D2 affinity switch model), while starting a high D2 affinity antipsychotic predicted an increase in symptoms (B = 3.29, P < .05 in D2 affinity switch model).
Conclusions: The results show that switching from an FGA to an SGA does not necessarily result in a reduction of TD or parkinsonism. Only stopping all antipsychotics reduces the severity of parkinsonism, and starting an FGA or a high D2 affinity antipsychotic may reduce the severity of TD.
Effect of Antipsychotic Type and Dose Changes
on Tardive Dyskinesia and Parkinsonism Severity
in Patients With a Serious Mental Illness:
The Curaçao Extrapyramidal Syndromes Study XII
ABSTRACT
Objective: To test the efficacy of current treatment recommendations for parkinsonism and tardive dyskinesia (TD) severity in patients with severe mental illness (SMI).
Methods: We present an 18-year prospective study including all 223 patients with SMI (as defined by the 1987 US National Institute of Mental Health, which were based on DSM-III-R diagnostic criteria) receiving care from the only psychiatric hospital of the former Netherlands Antilles. Eight clinical assessments (1992–2009) focused on movement disorders and medication use. Tardive dyskinesia was measured by the Abnormal Involuntary Movement Scale and parkinsonism by the Unified Parkinson’s Disease Rating Scale. Antipsychotics were classified into first-generation antipsychotic (FGA) versus second-generation antipsychotic (SGA) and high versus low dopamine 2 (D2) affinity categories. The effect that switching has within each category on subsequent movement scores was calculated separately by using time-lagged multilevel logistic regression models.
Results: There was a significant association between reduction in TD severity and starting/switching to an FGA (B = −3.54, P < .001) and starting/switching to a high D2 affinity antipsychotic (B = −2.49, P < .01). Adding an SGA to existing FGA treatment was associated with reduction in TD severity (B = −2.43, P < .01). For parkinsonism, stopping antipsychotics predicted symptom reduction (B = −7.76, P < .01 in FGA/SGA-switch model; B = −7.74, P < .01 in D2 affinity switch model), while starting a high D2 affinity antipsychotic predicted an increase in symptoms (B = 3.29, P < .05 in D2 affinity switch model).
Conclusions: The results show that switching from an FGA to an SGA does not necessarily result in a reduction of TD or parkinsonism. Only stopping all antipsychotics reduces the severity of parkinsonism, and starting an FGA or a high D2 affinity antipsychotic may reduce the severity of TD.
J Clin Psychiatry
https://doi.org/10.4088/JCP.16m11049
© Copyright 2017 Physicians Postgraduate Press, Inc.
aDepartment of Psychiatry and Psychology, Maastricht University Medical Centre, South Limburg Mental Health and Teaching Network, Maastricht, The Netherlands
bPsychiatric Centre GGz Centraal, Amersfoort, The Netherlands
cKing’s College London, King’s Health Partners, Department of Psychosis Studies, Institute of Psychiatry, London, United Kingdom
dPsychiatric Centre GGz Curaçao, Groot Kwartier, Curaçao
eParnassia Psychiatric Institute, The Hague, The Netherlands
fDepartments of Psychiatry and gNeurology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
hDepartment of Epidemiology, Columbia University, New York, New York
*Corresponding author: Charlotte L. Mentzel, MD, Innova, GGz Centraal, Utrechtseweg 266, 3818 EW Amersfoort, The Netherlands ([email protected]).
Despite the introduction of second-generation antipsychotics (SGAs), medication-induced movement disorders still occur frequently in psychiatric patients.1 Although movement disorders may have a lower incidence with SGAs compared with first-generation antipsychotics (FGAs), they are still highly prevalent side effects of antipsychotics.2–4 Movement disorders are especially prevalent among patients with serious mental illness (SMI), owing to frequent polypharmacy and because more severe symptoms are associated with increased risk of movement disorders.5–8
The 2 most prevalent movement disorders in this sample are parkinsonism and tardive dyskinesia (TD).1,9,10 Current treatment guidelines vary for the management of tardive dyskinesia.11–13 For parkinsonism, lowering antipsychotic dose is the recommended first step, followed by switching to a lower dopamine 2 (D2) affinity antipsychotic or an SGA.12,13 Randomized controlled trials on starting or switching antipsychotic medication mostly focus on the risk of developing movement disorders but not on the treatment of movement disorders. Even in large real-world trials, patients with treatment resistance or cognitive disorders (Clinical Antipsychotic Trials of Intervention Effectiveness [CATIE])8 or substance misuse (Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study [CUtLASS 1]),14 all of which represent known moderators of movement disorders,15 were excluded, reducing generalizability.
Given that lowering the antipsychotic dose or switching the antipsychotic are acknowledged therapeutic strategies for movement disorders, it is important to examine the effect of changing antipsychotic dose or type on the presence and severity of TD and parkinsonism. In a long-term naturalistic follow-up study with few exclusion criteria, findings can most easily be generalized to other settings. We, therefore, examined these issues over an 18-year period in a representative sample, consisting of all clinical psychiatric patients in a naturalistic well-defined catchment area, namely the islands of the former Netherlands Antilles. Our aims were to verify the efficacy of switching antipsychotic type or lowering antipsychotic dose on lowering parkinsonism and TD severity.
METHODS
Subjects
Data originated from a cohort of 223 patients hospitalized or receiving structured outpatient care from the only psychiatric hospital in the Netherlands Antilles, the Dr. D. R. Capriles Clinic. The Institutional Review Board approved the study, and informed consent was obtained from all patients included in the study. Over the course of 18 years, a total of 8 assessments focusing on movement disorders were carried out. More information about the design of the study can be found in a previous publication.16
Inclusion criteria were minimum age of 18 years and cumulative exposure to antipsychotics of at least 3 months; current antipsychotic use was not required. All patients met the US 1987 National Institute of Mental Health’s definition of SMI, which was based on DSM-III-R diagnostic criteria.17 The exclusion criterion was a history of neurologic disorders affecting motor function. Of the 223 patients that met the original inclusion criteria, a further 22 were later excluded because they had undergone a lobotomy prior to the study and, therefore, were not considered representative of current patients. Also, patients with dementia (n = 7) or mental retardation (n = 3) as primary diagnosis were excluded, although these disorders were not excluded when they were not the primary diagnosis. This resulted in a dataset of 191 patients (162 inpatients could be included at T0 in 1992; for practical considerations, the 29-day treatment patients on the island were included at T1 in 1993). The advantage of an island is that it is relatively easy to minimize the dropout rate, as patients rarely moved off the island and thus were easy to locate at the next time point; the main reason for attrition was death.
Movement disorder severity was assessed in a standardized manner, using the Abnormal Involuntary Movement Scale (AIMS)18 for TD and the Unified Parkinson’s Disease Rating Scale (UPDRS)19 for parkinsonism, administered by the same 2 raters (P.N.vH. and G.E.M.) at all 8 time points. Tardive dyskinesia was defined according to the Schooler and Kane criteria16,27; parkinsonism was defined as a score of at least 2 on a rigidity or tremor item or a score of at least 3 on a bradykinesia item.16 The first assessment was in 1992; the 7 subsequent measurements were in 1993, 1994, 1996, 1997, 1998, 2001, and 2009, respectively.
In addition, information on medication was collected by a trained physician. Information on age, DSM-III-R diagnosis, sex, and cocaine use was extracted from the patient’s file at the time of inclusion. Cocaine use was also assessed at the first 4 time points.
Definition and Coding of Variables
The defined daily dose (DDD) was calculated for both antipsychotics and benzodiazepines, and information on antipsychotic type and administration route was extracted.19,20 The total anticholinergic load of the combined medication was calculated for each patient by summing the anticholinergic load of each medication according to the Anticholinergic Drug Scale (ADS).20,21 The ADS assigns a score to each medication (ranging from 0 to 3) in accordance with the level of anticholinergic action of the compound.
Change scores were defined as the difference between the current and the previous assessment for (1) the AIMS or UPDRS and (2) antipsychotic DDD for antipsychotic dose change. For both TD and parkinsonism, an FGA/SGA switch, defined as starting and stopping an antipsychotic medication, switching from FGA to SGA, and adding an SGA to an FGA, was coded for all 8 time points. FGAs used in the study population were benperidol, droperidol, fluphenazine, flupentixol, haloperidol, promethazine, penfluridol, pericyazine, perphenazine, pimozide, pipamperon, thioridazine, trifluoperazine, and zuclopenthixol; SGAs used were clozapine, olanzapine, quetiapine, risperidone, and sulpiride. A separate analysis was done for both movement disorders based on D2 affinity. FGAs were coded as high D2 affinity antipsychotics, except for promethazine, which was coded as a low D2 affinity antipsychotic. SGAs were coded as low D2 affinity antipsychotics, except for risperidone, which was coded as a high D2 affinity antipsychotic. Hereafter, models that focus on switching from an FGA to an SGA will be referred to as FGA/SGA models, and models that focus on switching from a high D2 affinity antipsychotic to a lower D2 affinity antipsychotic will be referred to as D2 affinity models.
Statistical Analyses
The Stata22 XTMIXED command was used, given the multilevel structure with repeated assessments clustered within subjects. Per movement disorder, 3 analyses were carried out, 2 for the antipsychotic switch and 1 for the effect of antipsychotic dose change on TD and parkinsonism, respectively. In all analyses, the main independent variables were time lagged; all other independent variables were either time independent or pertaining to the current time point.
For TD and parkinsonism, the dependent variable was the difference over 2 consecutive time points in the AIMS and UPDRS scores, respectively. For both TD and parkinsonism switch analyses, the main independent variable was antipsychotic switch (either FGA/SGA switch or D2 receptor affinity switch in 2 different models). For the change in antipsychotic dose analyses, the main independent variable was the change in antipsychotic DDD over 2 consecutive time points.
All analyses were corrected for (1) age, sex, diagnosis, and cocaine use as time-independent variables and (2) benzodiazepine DDD, anticholinergic load, administration route of the antipsychotic, and antipsychotic DDD as time-dependent variables. For the antipsychotic dose change analysis, the main independent variable antipsychotic type was included instead of antipsychotic DDD as the time-dependent variable. All regression models were checked for a normal distribution of the residuals and the absence of heteroscedasticity.
RESULTS
At baseline, patients had a mean (SD) age of 50 (16) years; men (n = 139) had a mean age of 47 (14) years years and women (n = 52), a mean age of 58 (19) years. Nearly all patients (95.4%) were of African-Caribbean origin. The primary diagnoses according to DSM-III-R criteria were schizophrenia (80.2%), affective disorder (5.1%), and other (14.7%). Cocaine use was stable within patients over the time points, with 17% of patients using the drug.
Prevalence and Persistence of Tardive Dyskinesia and Parkinsonism
The mean prevalence for both movement disorders was high, with an average of 54% for TD and 35% for parkinsonism (Table 1); 18% of patients had both movement disorders. Both disorders showed a relapsing-remitting course with a mean persistence to the next time point of 70% (range, 67%–72% per time point) and 59% (range, 56%–72% per time point) for TD and parkinsonism, respectively. The results for the FGA/SGA models were very similar to those of the D2 affinity models.
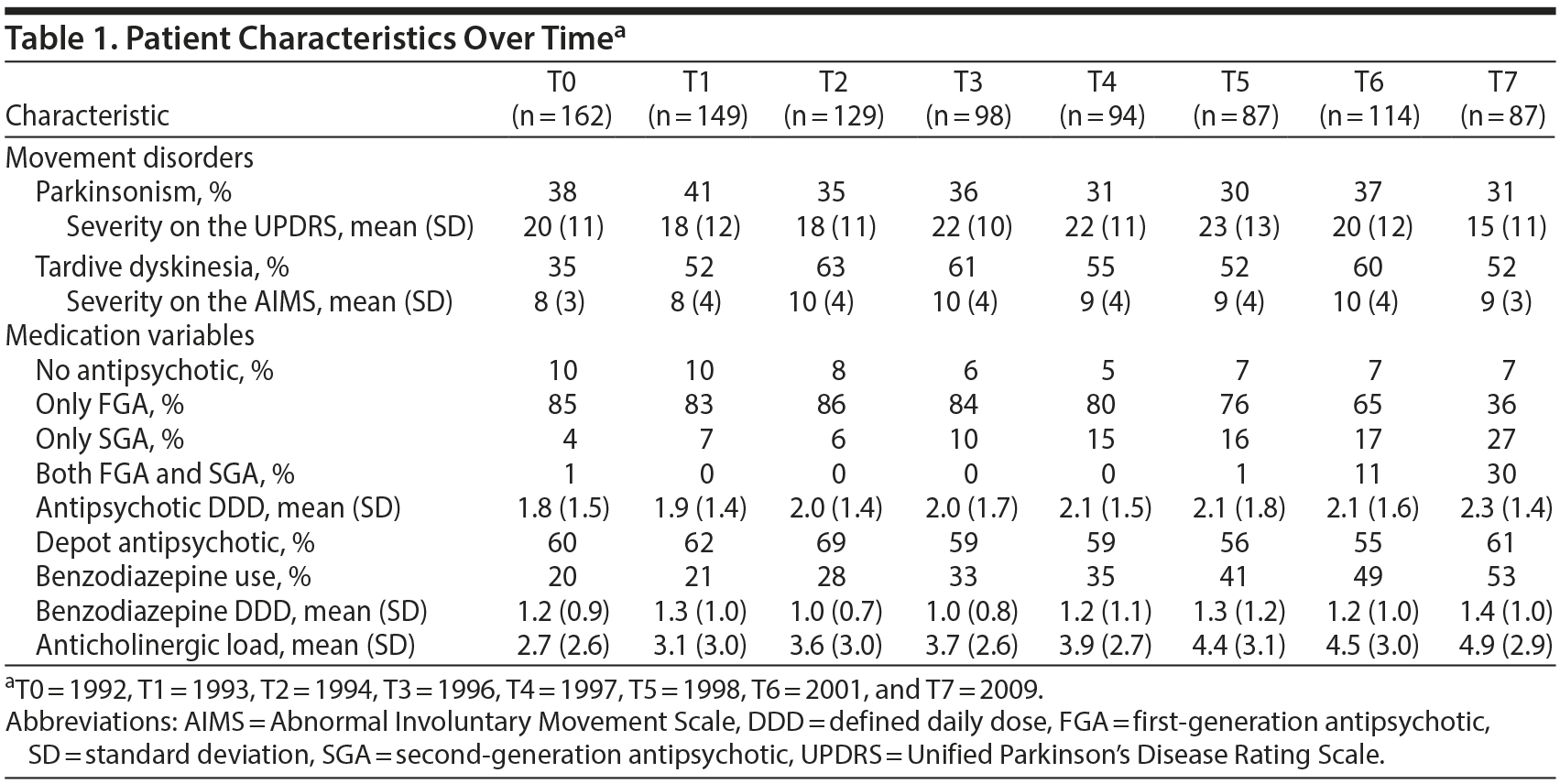
Tardive Dyskinesia
Patients with TD had a mean AIMS score of 9.1 (SD = 3.9). Both the FGA/SGA and the D2 affinity switch time-lagged multilevel logistic regression models yielded significant coefficients for switching to/starting an FGA or an antipsychotic with high D2 receptor affinity (B = −3.54, P < .001 and B = −2.49, P < .01, for the respective FGA/SGA and D2 receptor affinity switch models) (Table 2). In the FGA/SGA switch analysis, adding an SGA to existing FGA treatment also reduced TD severity (B = −2.43, P < .010).
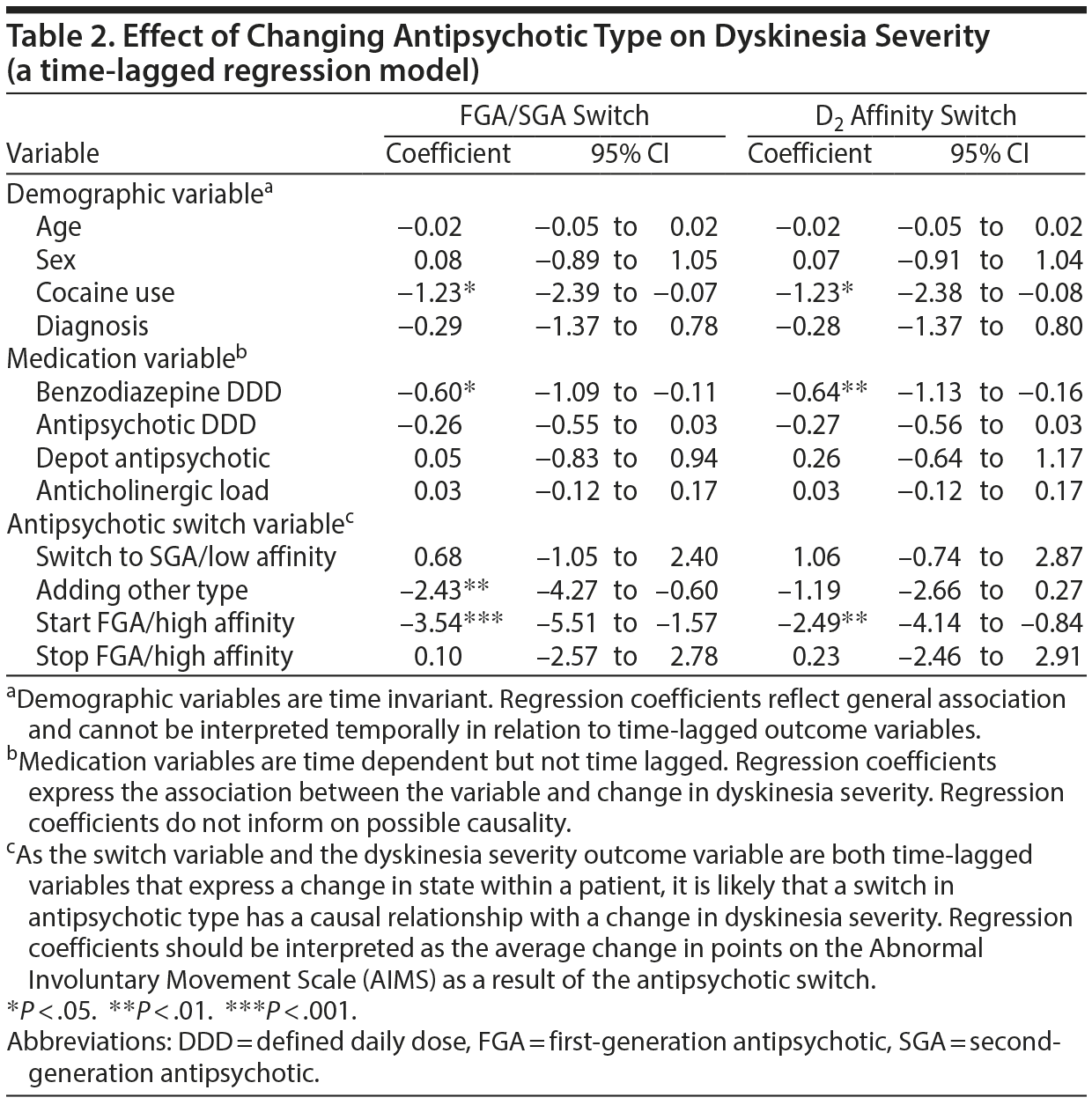
Mean antipsychotic DDD was 2.09 (1.81–2.32), with the mean DDD getting higher with each subsequent time point (Table 1). Increasing the antipsychotic dose by 1 DDD resulted in a reduction of TD severity of 0.42 points on the AIMS (P < .05). Tardive dyskinesia severity was also related to benzodiazepine DDD (B = −0.71, P < .01).
Parkinsonism
Patients with parkinsonism had a mean UPDRS score of 19.6 (SD = 11.6). Both the FGA/SGA and the D2 affinity switch time-lagged multilevel logistic regression models yielded significant coefficients for stopping antipsychotics, with a severity reduction of over 7 points (B = −7.76, P < .01 for the FGA/SGA model and B = −7.74, P < .01 for the D2 affinity model) (Table 3). In the D2 affinity model, starting antipsychotics in unmedicated patients was followed by an increase in parkinsonism severity (B = 3.29, P < .05).
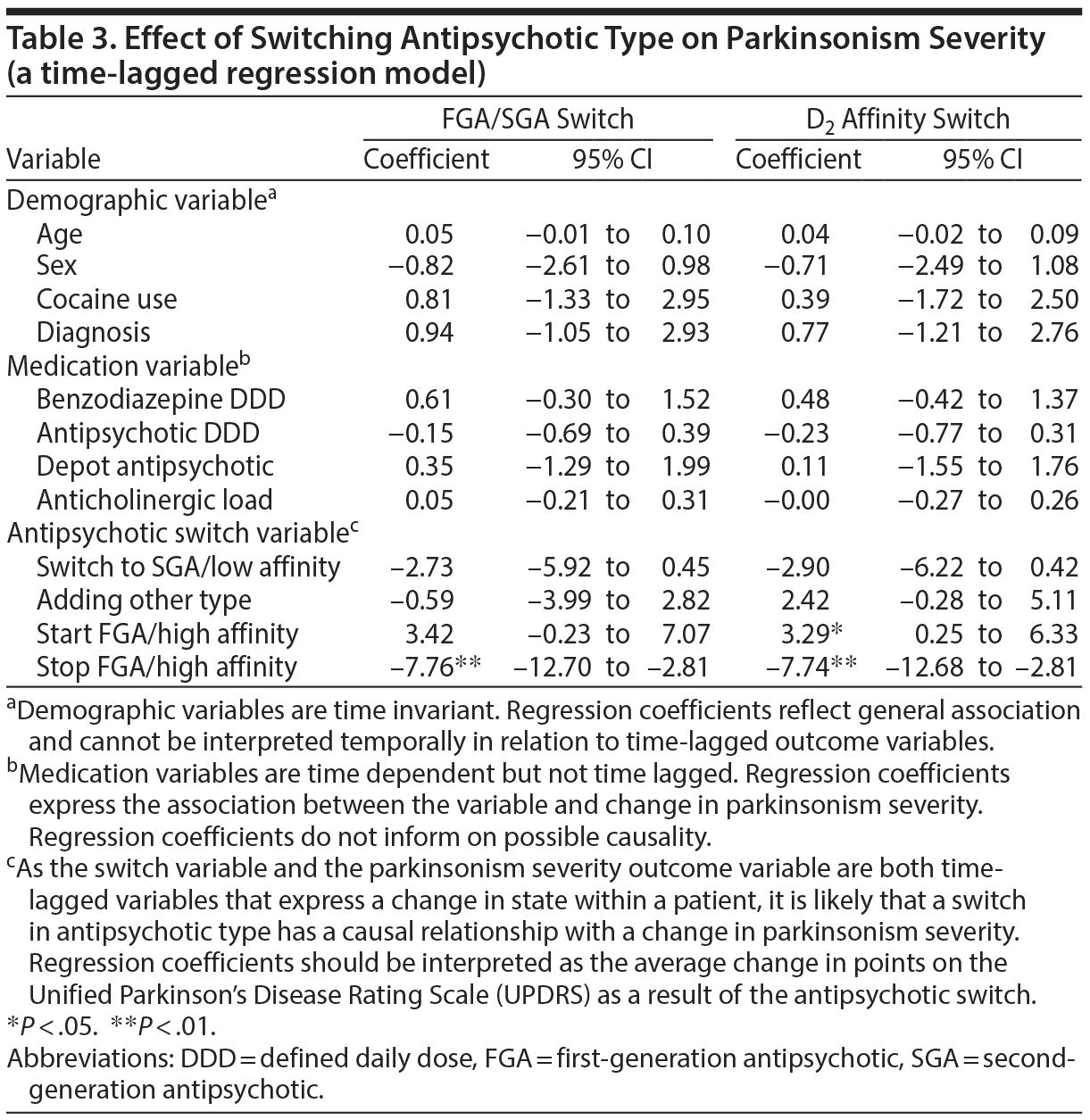
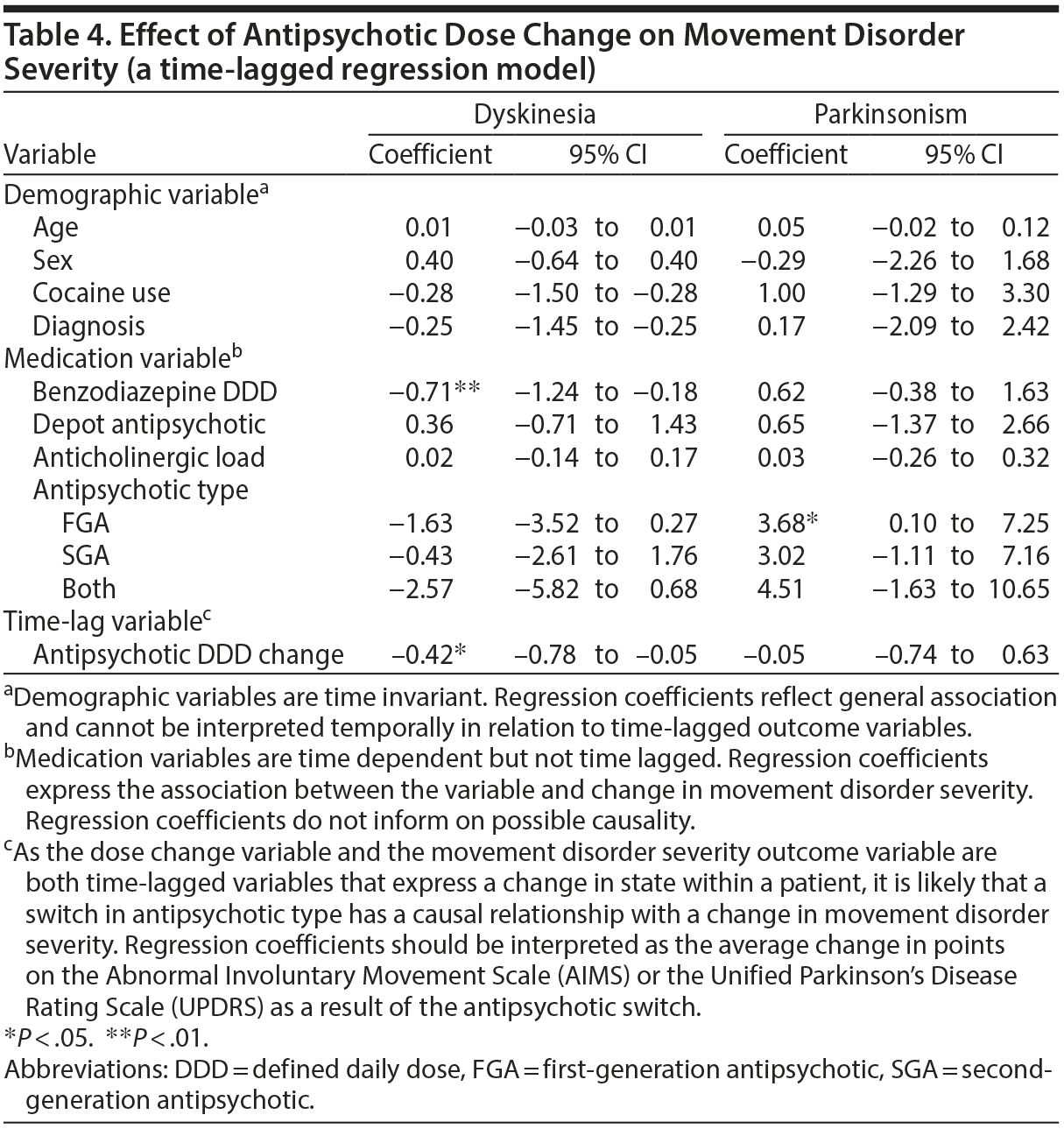
No significant association was found between changing antipsychotic dose and parkinsonism (B = −0.05; Table 4). There was, however, a significant association between higher parkinsonism severity and FGA use (B = 3.68, P < .05).
DISCUSSION
The results show that switching from an FGA to an SGA in patients with SMI does not necessarily lead to a reduction of dyskinetic or parkinsonism symptoms. In both the parkinsonism and TD analyses, switching from an FGA to an SGA or from a high D2 affinity antipsychotic to one with a lower affinity had no significant impact on the movement disorder. Instead, in the case of parkinsonism, only complete cessation of antipsychotic medication resulted in significantly lower UPDRS scores. In the TD analysis, adding an SGA to current FGA treatment or starting an FGA resulted in significantly lower scores. Surprisingly, lowering the antipsychotic dose had no effect on parkinsonism severity. One explanation is that movement disorders in patients with SMI behave differently than movement disorders in patients with less severe mental disorders. Therefore, more research on the treatment of movement disorders in patients with SMI is warranted.
Tardive Dyskinesia
According to English and American treatment guidelines, there is insufficient evidence for switching11,12 from an FGA to an SGA as treatment of TD. In contrast, the Dutch treatment guideline classifies switching as level-3 evidence.13 This guideline is based on 2 studies23,24 with the strongest evidence coming from the industry-sponsored trial.24 Our analysis, however, showed the opposite pattern, with a 3-point reduction on the AIMS for starting or switching to an FGA and a 2- to 3-point reduction for adding an SGA to FGA treatment. While a lower incidence of TD with SGA medication has been extensively documented,4 the treatment of established TD is difficult. Jeste and colleagues25 found remission of TD after cessation of all antipsychotic medication in 37% of cases. However, this study25 is over 30 years old. More recent studies show only a limited effect of antipsychotic dose reduction. Complete cessation of all antipsychotics leads to complete remission in only 2% of cases25,26 and to a reduction of severity in 20% cases. While it is possible that switching to an FGA reduces the probability of spontaneous remission of the TD, spontaneous remission is most unlikely given the 2% probability of remission with cessation of all antipsychotic medication.25,26
In our study, increasing antipsychotic dose led to a small but significant reduction of TD symptoms. This finding is in line with previous observations that an increase of the antipsychotic dose can mask TD.26,27 Adding an SGA to existing FGA treatment was also significantly associated with a TD severity reduction, which could be the same masking effect, but it is also possible that adding an SGA to an FGA truly reduces TD severity. The current study cannot distinguish between the 2 scenarios. However, antipsychotic polypharmacy has many disadvantages, among which is a sharp increase in the rate of side effects. These include parkinsonism, cognitive impairment, weight gain, and diabetes27,28; therefore, adding an SGA to an FGA cannot be recommended.
In all models, known moderators of movement disorders were added to improve efficiency of the statistical models. The time-dependent medication variables can be interpreted as having an association with a change in movement disorder outcome, but interpretations of causality cannot be made. In the TD analyses, benzodiazepine DDD was significantly associated with an increase in AIMS severity in both models. Time-independent variables such as cocaine use were added to improve the efficiency of the model; however, it is impossible to interpret any association between those variables and TD severity.
Parkinsonism
Current treatment guidelines suggest lowering the antipsychotic dose if parkinsonism occurs, with switching to an SGA as the second step.12,13 However, our current long-term follow-up study in patients with SMI does not support these interventions. While the few randomized controlled trials on treating parkinsonism in patients without SMI had small to moderate effect sizes,28–30 the association between antipsychotic type and dose and parkinsonism symptoms in patients with SMI has also been inconsistent.7,10 Possible reasons for this are that patients with SMI generally receive such a high dose of antipsychotic medication,1,30,31 around 2 DDD in our study, that a small dose reduction will have little effect. It is also possible that bradykinesia and parkinsonism symptoms in patients with SMI are not side effects of antipsychotic medication but neurologic soft signs that are part of the endophenotype of the underlying mental disorder.31,32 Whatever the reason, these results point to a difference between patients with SMI and patients without SMI, possibly warranting different treatment algorithms.
Strengths
Naturalistic studies, such as the Curaçao Extrapyramidal Syndromes Study, are important to test the results of pharmacologic interventions in a real-world setting. The Curaçao Extrapyramidal Symptoms Study is one of the few longitudinal studies in SMI patients with a long follow-up and many repeated measures, which makes it ideal for studying movement disorders, as these often show a relapsing and remitting course, in unique recurrent patterns, over the years. Because all patients with SMI in a well-defined catchment area were included, loss to follow-up was mainly due to death; and given the naturalistic design of the study, results can be considered representative for comparable real-world SMI populations. The lagged design also allows the study to examine the effect of switching antipsychotics on movement disorders at a later time point.
Another strongpoint is that, over the years, all ratings were carried out by the same raters, which reduced problems of interrater reliability.
Limitations
Some limitations need to be taken into account when interpreting the results. First, in this naturalistic study, the reason for switching antipsychotics is unknown and could not be included as a possible confounder in the analyses. Switching may occur for a variety of reasons, ie, development of movement disorder, lack of effect of the medication, or a request from patients or their family. These reasons could have influenced the results, as patients with a high risk of developing movement disorders might receive a different pharmacologic regimen compared to patients with a low risk. However, the time-lagged design, in which patients function as their own controls, limits this possible bias. If there is such a bias, it would be expected that regression coefficients would be more extreme but in the same direction. Second, the study population consisted mostly of participants who were of African-Caribbean origin; therefore, generalizing the results to other ethnic groups requires caution. However, Owens15 rightly states that differences in movement disorder risk in different ethnic groups are likely due to confounding between ethnicity and medication use. Third, the study could be underpowered due to the limited number of antipsychotic switches (40 switches to SGA; 36 switches to high D2 affinity), and the variables showed truncated distributions. Lack of statistical significance can therefore not be interpreted as lack of clinical effect. Instead, the size of regression coefficients should be considered when judging whether a change is clinically relevant. For example, in the antipsychotic switch and parkinsonism analyses, switching to an SGA/low D2 affinity antipsychotic has a large regression coefficient even though the confidence interval does contain the 0. Also, while there were no indications for heteroscedasticity, the residuals were symmetrical but not normally distributed. However, when assumptions were relaxed in bootstrapped analyses, very similar results were obtained. Therefore, we chose to present the original results instead of the bootstrap analyses.
Finally, in an ideal situation, we would have considered all antipsychotics on their individual merits instead of classifying them in 2 groups. As Leucht et al33 have shown, the characteristics of antipsychotics vary widely within the FGA and SGA groups. Leucht and colleagues33 therefore propose considering each individual antipsychotic on its own merits. However, considering all antipsychotics separately would leave our study underpowered. In order to assess the validity of the classic FGA/SGA classification while not sacrificing too much power, we replicated the analyses with antipsychotics classified according to their D2 receptor affinity.32,34 Also, patients in our study did not receive some of the newer antipsychotics such as aripiprazole; thus, no conclusions can be drawn about these antipsychotics.
In conclusion, our study suggests that movement disorders in patients with SMI may respond differently to switching antipsychotics than treatment guidelines would lead us to expect. Results indicate that, with the exception of stopping all antipsychotic medication for parkinsonism, effects of switching antipsychotic medication on both movement disorders are modest. However, there are additional considerations when it comes to clinical recommendations. Stopping medication increases risk of psychotic relapse, so stopping cannot be recommended for more than a brief interval to relieve parkinsonism. Moreover, some patients may have both parkinsonism and TD leading to conflicting treatment advice. For parkinsonism, if antipsychotic cessation is impossible, dose reduction may be attempted, as it has little clinical consequence. Switching antipsychotics to SGAs or low D2 affinity antipsychotics solely to reduce parkinsonism is not recommended, given the small effect and that these antipsychotics have their own limitations such as sedation, weight gain, and other metabolic and anticholinergic side effects. For TD, switching to an FGA may be a strategy to provide relief from TD in some cases. However, as it may increase parkinsonism or akathisia, this switch may not be wise, especially in elderly patients. An antipsychotic dose increase can also be considered, as it may mask the TD symptoms for at least a number of years, but it increases the risk of dose-dependent side effects. Adding an SGA to current FGA treatment cannot be recommended, because antipsychotic polypharmacy carries a much higher risk for psychiatric and somatic side effects.
Submitted: July 1, 2016; accepted January 3, 2017.
Online first: February 14, 2017.
Drug names: aripiprazole (Abilify), clozapine (Clozaril, FazaClo, and others), droperidol (Inapsine and others), olanzapine (Zyprexa and others), pimozide (Orap and others), promethazine (Phenergan, Remsed, and others), quetiapine (Seroquel and others), risperidone (Risperdal and others).
Potential conflicts of interest: In the past 5 years, the Maastricht University psychiatric research fund managed by Dr van Os has received unrestricted investigator-led research grants or recompense for presenting research at meetings from Lundbeck, Lilly, Janssen-Cilag, and Servier, companies that have an interest in the treatment of psychosis and depression. Dr Tijssen has received research grants from STW Technology society—NeuroSIPE, the Netherlands organization for scientific research—NWO Medium, Prinses Beatrix Spierfonds, Gossweiler Foundation, and Stichting Wetenschapsfonds Dystonie Vereniging and unrestricted grants from Ipsen, Allergan, and Medtronic for a dystonia nurse, DystonieNet, and a teaching course. Drs Mentzel, Bakker, Drukker, Matroos, Hoek, and van Harten report no conflicts of interest.
Funding/support: The research was supported by a grant from the National Antilles Foundation for Clinical Higher Education (NASKHO). There is no grant number associated with the grant.
Role of the sponsor: NASKHO, the study sponsor, is a foundation that stimulates education and research on the previous Netherlands Antilles. They had no role in design or conduct of the study; collection, management, analysis, or interpretation of the data; or preparation, review, or approval of the manuscript.
REFERENCES
1. Bakker PR, de Groot IW, van Os J, et al. Long-stay psychiatric patients: a prospective study revealing persistent antipsychotic-induced movement disorder. PLoS One. 2011;6(10):e25588. PubMed doi:10.1371/journal.pone.0025588
2. Correll CU, Schenk EM. Tardive dyskinesia and new antipsychotics. Curr Opin Psychiatry. 2008;21(2):151–156. PubMed doi:10.1097/YCO.0b013e3282f53132
3. Attard A, Taylor DM. Comparative effectiveness of atypical antipsychotics in schizophrenia: what have real-world trials taught us? CNS Drugs. 2012;26(6):491–508. PubMed doi:10.2165/11632020-000000000-00000
4. Leucht S, Corves C, Arbter D, et al. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373(9657):31–41. PubMed doi:10.1016/S0140-6736(08)61764-X
5. Tenback DE, van Harten PN, Slooff CJ, et al; SOHO Study Group. Worsening of psychosis in schizophrenia is longitudinally associated with tardive dyskinesia in the European Schizophrenia Outpatient Health Outcomes study. Compr Psychiatry. 2007;48(5):436–440. PubMed doi:10.1016/j.comppsych.2007.05.003
6. van Harten PN, Bakker PR, Mentzel CL, et al. Movement disorders and psychosis, a complex marriage. Front Psychiatry. 2015;5:190. PubMed doi:10.3389/fpsyt.2014.00190
7. Bakker PR, de Groot IW, van Os J, et al. Predicting the incidence of antipsychotic-induced movement disorders in long-stay patients: a prospective study. Epidemiol Psychiatr Sci. 2013;22(4):375–379. PubMed doi:10.1017/S204579601300019X
8. Miller DD, McEvoy JP, Davis SM, et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophr Res. 2005;80(1):33–43. PubMed doi:10.1016/j.schres.2005.07.034
9. Van Harten PN. Movement disorders associated with neuroleptics: the Curaçao Extrapyramidal Syndromes Study [dissertation]. Utrecht, The Netherlands: Utrecht University; 1998.
10. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127–148, viii. PubMed doi:10.1016/j.ncl.2010.10.002
11. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463–469. PubMed doi:10.1212/WNL.0b013e31829d86b6
12. Psychosis and Schizophrenia in Adults: Treatment and Management. NICE UK Web site. https://www.nice.org.uk/guidance/cg178. 2014.
13. van Alphen C, Ammeraal M, Blanke C, et al. Multidisciplinaire richtlijn schizofrenie. Utrecht, the Netherlands: Nederlandse Vereniging voor Psychiatrie; 2012.
14. Jones PB, Barnes TR, Davies L, et al. Randomized controlled trial of the effect on quality of life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (CUtLASS 1). Arch Gen Psychiatry. 2006;63(10):1079–1087. PubMed doi:10.1001/archpsyc.63.10.1079
15. Owens DC. A Guide to the Extrapyramidal Side-Effects of Antipsychotic Drugs. 2nd edition. New York, NY: Cambridge University Press; 2014. doi:10.1017/CBO9781139149112
16. van Harten PN, Matroos GE, Hoek HW, et al. The prevalence of tardive dystonia, tardive dyskinesia, parkinsonism and akathisia: the Curaçao Extrapyramidal Syndromes Study, I. Schizophr Res. 1996;19(2–3):195–203. PubMed doi:10.1016/0920-9964(95)00096-8
17. Ruggeri M, Leese M, Thornicroft G, et al. Definition and prevalence of severe and persistent mental illness. Br J Psychiatry. 2000;177:149–155. PubMed doi:10.1192/bjp.177.2.149
18. Lane RD, Glazer WM, Hansen TE, et al. Assessment of tardive dyskinesia using the Abnormal Involuntary Movement Scale. J Nerv Ment Dis. 1985;173(6):353–357. PubMed doi:10.1097/00005053-198506000-00005
19. Fahn S, Elton RL, Members of the UPDRS Development Committee. Unified Parkinson’s Disease Rating Scale. In: Fahn S, Marsden CD, Calne DB, Goldstein M, eds. Recent Developments in Parkinson’s Disease, Vol 2. Florham Park, NJ: Macmillan Health Care Information; 1987:153–164.
20. Collaborating Centre for Drugs Statistics Methodology. WHO Web site. http://www.whocc.no/atcddd/. Cited 2013 June.
21. Carnahan RM, Lund BC, Perry PJ, et al. The Anticholinergic Drug Scale as a measure of drug-related anticholinergic burden: associations with serum anticholinergic activity. J Clin Pharmacol. 2006;46(12):1481–1486. PubMed doi:10.1177/0091270006292126
22. Stata Statistical Software: Release 12 [computer program]. College Station, TX: StataCorp LP; 2011.
23. Tenback DE, van Harten PN, Slooff CJ, et al; SOHO Study Group. Effects of antipsychotic treatment on tardive dyskinesia: a 6-month evaluation of patients from the European Schizophrenia Outpatient Health Outcomes (SOHO) Study. J Clin Psychiatry. 2005;66(9):1130–1133. PubMed doi:10.4088/JCP.v66n0907
24. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696–701. PubMed doi:10.4088/JCP.v65n0516
25. Jeste DV, Wyatt RJ. Therapeutic strategies against tardive dyskinesia: two decades of experience. Arch Gen Psychiatry. 1982;39(7):803–816. PubMed doi:10.1001/archpsyc.1982.04290070037008
26. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166–176. PubMed doi:10.1007/s13311-013-0222-5
27. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486–487. PubMed doi:10.1001/archpsyc.1982.04290040080014
28. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527–542. PubMed doi:10.1517/14740338.2012.683523
29. Ritchie CW, Chiu E, Harrigan S, et al. The impact upon extra-pyramidal side effects, clinical symptoms and quality of life of a switch from conventional to atypical antipsychotics (risperidone or olanzapine) in elderly patients with schizophrenia. Int J Geriatr Psychiatry. 2003;18(5):432–440. PubMed doi:10.1002/gps.862
30. Cortese L, Caligiuri MP, Williams R, et al. Reduction in neuroleptic-induced movement disorders after a switch to quetiapine in patients with schizophrenia. J Clin Psychopharmacol. 2008;28(1):69–73. PubMed doi:10.1097/jcp.0b013e318160864f
31. Suzuki T, Uchida H, Tanaka KF, et al. Reducing the dose of antipsychotic medications for those who had been treated with high-dose antipsychotic polypharmacy: an open study of dose reduction for chronic schizophrenia. Int Clin Psychopharmacol. 2003;18(6):323–329. PubMed doi:10.1097/00004850-200311000-00003
32. Bachmann S, Degen C, Geider FJ, et al. Neurological soft signs in the clinical course of schizophrenia: results of a meta-analysis. Front Psychiatry. 2014;5:185.
33. Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951–962. PubMed doi:10.1016/S0140-6736(13)60733-3
34. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull. 2009;35(3):549–562. PubMed doi:10.1093/schbul/sbp006
This PDF is free for all visitors!





