Dementia is a clinical syndrome commonly encountered in clinical practice. Early onset cognitive impairment is always of particular concern and warrants further workup for diagnostic clarity, potentially reversible causes, and prognosis. Unfortunately, although there is increasing knowledge of certain patterns of early onset neurodegenerative disorders, such as early onset Alzheimer’s disease and frontotemporal dementia, more atypical cases exist that do not fit these molds. Here, the case is presented of a 67-year-old man who first developed cognitive impairment at age 47 years. He also had a history of hyperlipidemia, hypogonadism, Lyme disease, anxiety, and attention-deficit disorder. He developed executive function deficits, impaired concentration, apathy, and gait disturbance, which led to loss of job, reduction in household responsibilities, and social isolation. He underwent extensive neuropsychiatric workup and several treatment attempts (for Lyme disease and attention-deficit disorder) over the subsequent 20 years; however, he had progressive indolent neurocognitive decline. This workup ruled out known causes of neurodegeneration but was consistent with early onset atypical parkinsonism with dementia of unspecified etiology. This case demonstrates the course of an early onset dementia that, despite exhaustive medical workup, remains diagnostically unclear. This scenario is common across medical specialties, although not often written about. This article synthesizes the individual approaches of neurologists, psychiatrists, radiologists, infectious disease specialists, and psychologists when presented with the same case and the effective multidisciplinary integration of these efforts even when the exact diagnosis remains unknown.
Abstract
Dementia is a clinical syndrome commonly encountered in clinical practice. Early onset cognitive impairment is always of particular concern and warrants further workup for diagnostic clarity, potentially reversible causes, and prognosis. Unfortunately, although there is increasing knowledge of certain patterns of early onset neurodegenerative disorders, such as early onset Alzheimer’s disease and frontotemporal dementia, more atypical cases exist that do not fit these molds. Here, the case is presented of a 67-year-old man who first developed cognitive impairment at age 47 years. He also had a history of hyperlipidemia, hypogonadism, Lyme disease, anxiety, and attention-deficit disorder. He developed executive function deficits, impaired concentration, apathy, and gait disturbance, which led to loss of job, reduction in household responsibilities, and social isolation. He underwent extensive neuropsychiatric workup and several treatment attempts (for Lyme disease and attention-deficit disorder) over the subsequent 20 years; however, he had progressive indolent neurocognitive decline. This workup ruled out known causes of neurodegeneration but was consistent with early onset atypical parkinsonism with dementia of unspecified etiology. This case demonstrates the course of an early onset dementia that, despite exhaustive medical workup, remains diagnostically unclear. This scenario is common across medical specialties, although not often written about. This article synthesizes the individual approaches of neurologists, psychiatrists, radiologists, infectious disease specialists, and psychologists when presented with the same case and the effective multidisciplinary integration of these efforts even when the exact diagnosis remains unknown.
Prim Care Companion CNS Disord 2020;22(4):19nr02570
To cite: Newhouse A, Buch K, Chemali Z. Dementia unspecified: a multidisciplinary approach. Prim Care Companion CNS Disord. 2020;22(4):19nr02570.
To share: https://doi.org/10.4088/PCC.19nr02570
© Copyright 2020 Physicians Postgraduate Press, Inc.
aDepartment of Psychiatry, Massachusetts General Hospital, Boston, Massachusetts
bDepartment of Medicine, Massachusetts General Hospital, Boston, Massachusetts
cHarvard Medical School, Boston, Massachusetts
dDepartment of Neuroradiology, Massachusetts General Hospital, Boston, Massachusetts
eDepartment of Neurology, Massachusetts General Hospital, Boston, Massachusetts
*Corresponding author: Amy Newhouse, MD, Department of Psychiatry, Massachusetts General Hospital, WAC 812, 15 Parkman St, Boston, MA 02114 ([email protected]).
The diagnostic workup of early onset dementias often takes a circuitous route. We present the case of man who began developing symptoms of a neurodegenerative disorder with atypical parkinsonism at age 47 years. We summarize the evolution of his diagnostic workup, and in doing so, review current clinically available data to assess neurodegenerative disorders. Despite exhaustive efforts, his diagnosis remains unclear to date. This situation is common in clinical practice, though infrequently discussed. Here, we take the opportunity to demonstrate how clinically meaningful information can still be gleaned from an inconclusive diagnostic workup using a longitudinal and multidisciplinary approach.
CASE REPORT
History
Mr A is a 67-year-old, right-handed white man with a history of hyperlipidemia, hypogonadism, Lyme disease, anxiety, and attention-deficit disorder (diagnosed as an adult) who presented for a neuropsychiatric evaluation of cognitive, behavioral, and emotional changes, which began 20 years prior. Mr A was married, had 4 children, and had worked as a chemical engineer. At age 47 years, he began having difficulty with concentration, organizing, and planning. He could no longer prioritize his tasks at work or manage his schedule. His secretary had helped him compensate; however, after she left employment, he was unable to successfully function independently. He subsequently lost his job and despite multiple attempts was unable to secure new employment. At that time, he and his wife attributed his symptoms to the stress of raising 4 children and also caring for multiple ill family members. However, as these demands settled, his symptoms persisted. In addition to the cognitive impairment, he suffered from significant apathy and began socially isolating. When describing her husband, his wife said, “the lights went out.”
Mr A retained the ability to perform instrumental activities of daily living, such as driving, shopping, and cooking, and was able to bathe, dress, and feed himself. His math skills had significantly diminished, and his home finances were transitioned to online autopay. Keeping track of his schedule remained a major hurdle. He endorsed word-finding difficulty. His wife added that he had decreased understanding of abstract concepts and certain implied social innuendos and displayed mental rigidity.
Mr A denied experiencing depression or anhedonia, although he acknowledged mild anxiety, which he associated with his functional impairment. He had no symptoms of mania, psychosis, hallucinations, or posttraumatic stress disorder during this period. He had no sleep disturbance. He denied any motor symptoms, although his wife noted that his posture had become slightly stooped, his gait was shuffled, and he had loss of fine coordination.
There were many discrepancies between his subjective symptoms and his wife’s observations. However, both reported that the timeline of his symptoms began abruptly at age 47 years and had not progressed or improved since. They were also both convinced that the symptoms were due to Lyme disease.
Mr A’s family history was notable for late-onset dementia (unknown subtype) in a maternal aunt and multiple myeloma in his father. He had never smoked and denied any history of drug use. He drank, on average, 2 glasses of wine per week.
Examination
Mr A generally spoke only when asked direct questions and used simple, concrete, and often vague language. His speech was slow and soft with increased latency and occasional stutter. He demonstrated mild psychomotor retardation. His thought processes were logical though concrete, and he was unable to follow mildly complex instructions such as tracking a finger with his eyes while holding his head still. His eye movements were normal. He had increased tone in his upper extremities (left greater than right). His posture was stooped, but his stance was steady. He had reduced stride length and arm swing (left greater than right). Toe, heel, and tandem walk provoked posturing. The remainder of his examination was otherwise unremarkable.
Workup
Over the course of the past 20 years, Mr A had undergone an extensive workup at various locations throughout the state of Massachusetts. This workup included laboratories, electroencephalograms (EEGs), structural imaging, functional imaging, and neuropsychological testing.
Laboratories. Mr A’s laboratory work included normal complete blood count, comprehensive metabolic panel, thyroid-stimulating hormone, vitamin D, vitamin B12, methylmalonic acid, HIV, syphilis, eastern equine encephalitis antibody, bartonella antibody, borrelia miyamotoi antibody, brucella antibody, serum autoimmune encephalitis panel, heavy metal screen, and cerebrospinal fluid studies, including amyloid β (Aβ) and tau ratios (Aβ42: 824.95 pg/mL, t-tau: 118.45 pg/mL, p-tau: 26.2 pg/mL, Aβ tau index [ATI]: 2.17). He was found to have Lyme serology consistent with active disease (positive Lyme IgM antibodies and Lyme C6 peptide) in September 2017 at age 66 years. Erythrocyte sedimentation rate, C-reactive protein, thyroid peroxidase antibody, and homocysteine results were within normal limits. The antinuclear antibody test was positive: 1:40 (speckled pattern). The apolipoprotein E (ApoE) panel was heterozygous for ApoE3 and ApoE4.
Neuroimaging. Multiple magnetic resonance images (MRIs) had been performed over the past 20 years. We had access to MRIs from February 2006, October 2006, and January 2016. Imaging was performed with a contrast-enhanced MRI in conjunction with acquisition of vascular imaging of the intracranial and extracranial arteries. Pertinent negative findings on the MRI examination of the brain included a lack of abnormal intracranial enhancement, no intracranial mass lesions, no evidence of recent or sequela of remote infarctions, and no evidence of intracranial hemorrhage.
Mild, diffuse brain parenchymal volume loss was appreciated, but there was no significant regional pattern of volume loss, including the mesial temporal lobes, hippocampi, brain stem, and cerebellum. Somewhat disproportionate volume loss was seen involving the left superior parietal lobule, which had slowly progressed since the early MRI performed in 2006. Minimal punctate, nonenhancing, and scattered foci of T2/fluid-attenuated inversion recovery (FLAIR) signal hyperintensities were appreciated within the subcortical white matter of both cerebral hemispheres (a total of 3 lesions), which was also new since the prior MRI from 2006 (Figures 1 and 2).
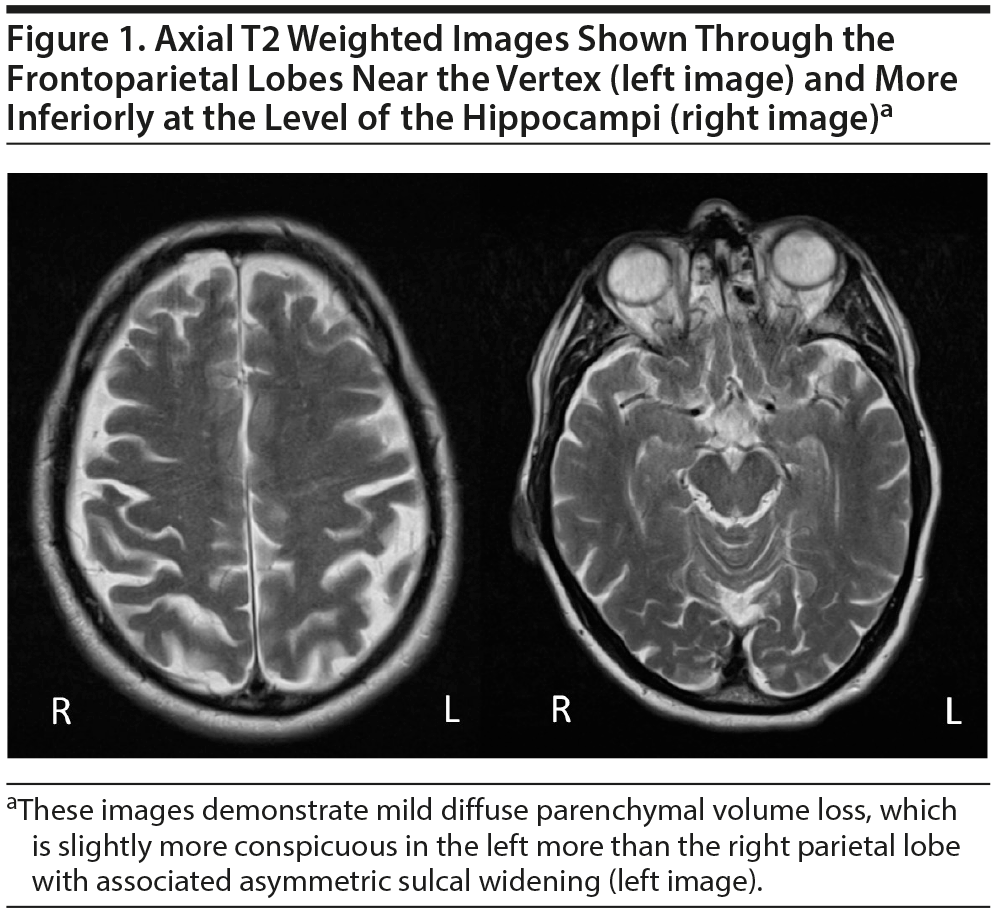
Vascular imaging demonstrated moderate focal stenosis involving the mid portion of the right cervical vertebral artery and minimal stenosis at the origin of the right cervical internal carotid artery. Intracranially, no hemodynamically significant stenosis or vascular malformation was noted.
He also had 2 positron emission tomography (PET) scans from October 2006 and April 2018 and 2 single-photon emission computerized tomography (SPECT) scans from September 2003 and June 2004. The PET scans showed mild patchy hypometabolism in the parietal lobes bilaterally. Normal physiologic uptake was observed in the bifrontal lobes, temporal lobes, occipital lobes, and cingulate gyrus bilaterally. The first SPECT scan showed moderate perfusion defects in the left temporal and occipital lobes and mild defects in the right parietal lobe; the scan a year later showed heterogeneous decreased uptake in the frontal and frontoparietal regions (right side greater than the left).
An EEG early in the course of Mr A’s illness showed bitemporal occasional theta wave activity, and another EEG 10 years later showed intermittent left temporal slowing. No epileptiform activity was ever detected.
Within a research protocol, Mr A had amyloid and tau PET scans that were read as borderline high, consistent with normal aging and not consistent with symptomatic Alzheimer’s disease (AD) (Figure 3). The MK-6240 tracer was bound only to the medial temporal lobes, consistent with Braak I/II staging. The PiB binding was nonspecific. A DAT (dopamine transporter) SPECT scan was unavailable.
Neuropsychological testing. Seven rounds of neuropsychological testing were undertaken between 1991 and 2015. Comparisons showed a drop in IQ from 135 (99th percentile) to 94 (35th percentile). The areas with significant impairments included reasoning, abstraction, hypothesis testing, auditory attention span, and emotion recognition. He had, to a lesser degree, deficits in working memory, visual processing speed, sustained and shifting attention, and spatial planning and organization. Although areas of phonemic fluency, single word reading, naming, and memory were relatively preserved, he scored in the average to low average range of performance in these domains, which most likely represented an overall decline from his premorbid level of functioning. Table 1 provides the 2015 testing results with prior comparisons where available.
At our initial evaluation, Addenbrooke’s Cognitive Examination Revised16 total score was 65/100 (16/18 in attention and orientation, 11/26 in memory, 4/14 in fluency, 23/26 in language, 11/16 in visuospatial). Of note, normative values are based on 63 controls aged 52–75 years and 142 dementia patients aged 46–86 years; a cutoff < 82 gives 84% sensitivity and 100% specificity for dementia.
Treatment History
Mr A had tried various medications over this time period to target mood, memory, and attention, all with little effect. These medications included citalopram, venlafaxine, bupropion, donepezil, memantine, methylphenidate, dextroamphetamine, atomoxetine, clonidine, and carbidopa/levodopa. The antidepressants, stimulants, and carbidopa/levodopa all caused bruxism. None led to any significant benefit. He also received treatment with 2 months of intravenous ceftriaxone for Lyme disease. While his laboratory results demonstrated resolution of potential infection, there was no associated cognitive improvement.
MULTIDISCIPLINARY CONFERENCE DISCUSSION
This case was presented and discussed at a multidisciplinary conference that included general neurologists and psychiatrists, behavioral neurologists and neuropsychiatrists, and a neuroradiologist. In addition, it included evaluations done by a neuroinfectious disease specialist and multiple neuropsychologists. The following discussion encapsulates the highlights of this collaboration.
In summary, Mr A is a 67-year-old right-handed man who presented for a neuropsychiatric evaluation with a 20-year history of cognitive impairment and apathy, in the absence of subjective reports of depression or severe anxiety, leading to inability to work or function at his baseline as of age 47 years. There was a disconnect between the subjective report of symptoms (that they had not progressed) and the objective neuropsychological data (which did show progressive deficits). The patient and his wife had latched on to the diagnosis of Lyme disease, a common scenario given the broad range of potential neuropsychiatric symptoms that can be seen in Lyme disease. In this case, however, it had been successfully treated without associated cognitive benefit.
His general neurologic examination demonstrated parkinsonism with anhedonia, apathy, bradyphrenia, and gait disturbance. His treatment history was notable for a pattern of developing the adverse effect of bruxism in response to medications with no tangible benefit otherwise. Taken together, his examination and treatment history were suggestive of a dopamine deficit.17
His testing demonstrated a progressive decline in his IQ from a level of superior intelligence to the 35th percentile, with deficits in attention and executive impairment. From a neurocircuitry perspective, his deficits pointed toward disruption of the frontal circuits. The dorsolateral prefrontal cortex is thought to be involved in working memory18 and cognitive flexibility,19 while the ventrolateral and orbital prefrontal cortices are involved in emotional processing and learning.20,21 Mr A demonstrated deficits in all of these realms.
From a neuroimaging perspective, the salient findings were mild diffuse brain parenchymal volume loss with disproportionate involvement of the left parietal lobe. These findings were seen in the absence of parenchymal signal abnormality and infratentorial volume loss. There was slight progression in atrophy over time. The PET scans showed mild patchy hypometabolism in the parietal lobes bilaterally. The SPECT scans initially showed moderate perfusion defects in the left temporal and occipital lobes and mild defects in the right parietal lobe but a year later showed heterogeneous defects in the frontal and frontoparietal regions bilaterally, though the right side was greater than the left. The amyloid and tau PET scans were consistent with normal aging.
Taken altogether, Mr A was felt to have early onset atypical (before age 65 years) parkinsonism with dementia of unspecified etiology. We hypothesized that his syndrome may reflect either a tauopathy or a α-synucleinopathy. α-synuclein is an intracellular protein that is present in the central and peripheral nervous system. Pathological aggregates within neurons are thought to be relevant to diseases such as Parkinson’s disease, dementia with Lewy bodies, multisystem atrophy, and pure autonomic failure.22 Disrupted dopamine signaling is felt to contribute to many of the symptoms in these diseases.22 Tau is an intracellular and extracellular protein, also present in the central and peripheral nervous system, that is relevant for stabilization of the axonal cytoskeleton. Intracellular pathological aggregates, known as neurofibrillary tangles, are thought to be relevant to diseases such as AD, progressive supranuclear palsy, frontotemporal dementia, parkinsonism linked to chromosome 17, corticobasal degeneration, postencephalitic parkinsonism primary age-related tauopathy, and familial multiple systems tauopathy with presenile dementia.23,24
Regional patterns of brain parenchymal volume loss on MRI can help distinguish between neurodegenerative disorders. A tauopathy classically has disproportionate volume loss that may involve 1 or both parietal lobes, 1 or both temporal lobes, and 1 or both hippocampi. Disproportionate volume loss involving the frontotemporal lobes would be more suggestive of a frontotemporal lobar degeneration. A combination of asymmetric cortical atrophy involving the superior parietal lobule and perirolandic regions, atrophy of the basal ganglia, and atrophy of the corpus callosum can be seen in the setting of corticobasal degeneration.25 Brain stem atrophy with predilection for the midbrain may be seen in the setting of a progressive supranuclear palsy with development of a “hummingbird brain stem.”26
Synucleinopathies can also manifest with variable patterns of brain parenchymal volume loss with a predilection for disproportionate volume loss of the frontal, parietal, and temporal lobes, as well as focal atrophy involving the midbrain and hypothalamus.27 In some types of synucleinopathies, such as multisystem atrophy disorders, T2 signal hyperintensity may be present in the pontocerebellar tracks and putamen, along with volume loss favoring these structures.28,29 Parkinson’s disease manifests with T1 signal hyperintensity within the regions of the substantia nigra and loss of normal susceptibility signal intensity within the substantia nigra.
Given the low number of T2/FLAIR hyperintense foci, relative peripheral location of these foci, and absence of associated encephalomalacia, blood products, diffusion signal abnormality, and enhancement in these locations, these findings were felt to be very nonspecific and likely related to chronic microvascular disease. While these findings are nonspecific, specific attention was given to these T2/FLAIR signal hyperintensities considering the patient’s positive Lyme serologies. In the acute and subacute settings, neuroborreliosis may manifest as a meningoencephalitis with enhancement of the leptomeninges and cranial nerves. If the syndrome progresses to encephalomyelitis, tumefactive lesions can be seen throughout the brain. More commonly, multiple small, patchy, nonenhancing T2/FLAIR hyperintense lesions develop throughout the brain parenchyma, predominately involving the subcortical and juxtacortical white matter, as well as at the callosal-septal interface. MRI findings in our patient were not suggestive of neuroborreliosis secondary to the low number of T2/FLAIR lesions and lack of juxtacortical and callosal-septal lesions.30,31 Additionally, central Lyme disease can also cause atrophy specific to the temporal or diffuse cortical hypometabolism, neither of which were seen in this patient.
Mr A’s history was not consistent with the more commonly defined phenotypes, such as AD or Parkinson’s disease. The less distinct synucleinopathies and tauopathies can have diverse presentations and co-occur. CSF biomarkers were not consistent with AD in that his Aβ42, t-tau, and p-tau levels were all within the normal ranges. Values consistent with AD would include decreased levels of Aβ42 and increased levels of both t-tau and p-tau, leading to a reduced ATI.32 While these biomarkers cannot diagnose AD alone, all 3 within normal range rules out AD.32 These biomarkers can distinguish between AD and non-AD dementias; however, they cannot confirm another type.32 At this point, our differential includes multiple systems tauopathy with presenile dementia, microtubule associated protein tau mutation carrier and frontotemporal dementia linked to parkinsonism-17, and TAR DNA-binding protein 43 related to C9ORF72 mutation or progranulin.33,34 These disorders can have an abrupt onset followed by a relatively subacute course, such as that of Mr A. The FDG hypometabolism in the bilateral temporal lobes, medial parietal, and frontal cortices seen in the nuclear studies also support this differential.35–37 We could potentially provide further diagnostic information if a dopamine PET scan was available and if the patient agreed to genetic testing. He did participate in speech and cognitive therapy, which both he and his wife found useful. We plan to continue to monitor Mr A longitudinally.
CONCLUSION
In conclusion, this case exemplifies the course of an early onset dementia that, despite exhaustive medical workup, remains diagnostically unclear. This scenario is common across medical specialties and while often not written about is an important topic to discuss. Despite no clear pathophysiologic diagnosis to account for his clinical syndrome, the diagnostic workup did produce several high-yield findings. The lack of response to antibiotic treatment combined with neuroimaging allowed us to rule out Lyme disease as the primary etiology. The normal CSF biomarkers ruled out AD. His symptoms combined with the adverse reactions to treatment led us to theorize the relevant neurocircuitry and neurotransmitter involved. The structural and functional neuroimaging did confirm evidence of atrophy suggestive of neurodegeneration. Taken together, this information allowed us to generate a differential for neurodegenerative disorders not as commonly encountered and not yet well understood. This case highlights the importance of serial monitoring over time. Situations like this, which while potentially frustrating in real time, do inform future care by driving research and adding another documented history for reference. We feel this case also demonstrates the benefit of and necessity for a collaborative, multidisciplinary approach to care, especially when facing complex cases with no clear answers in sight.
Clinical Points
- Complex neurocognitive presentations for which a diagnosis remains unclear despite exhaustive workup are best monitored with serial evaluations over time and a multidisciplinary approach.
- Neurodegenerative disorders can have broad symptomatology; thinking through the individual symptoms (such as apathy, anhedonia, bradyphrenia and parkinsonism) can help identify relevant neurocircuitry.
- Regional patterns of brain parenchymal volume loss on magnetic resonance imaging can help distinguish between neurodegenerative disorders.
- Diagnoses should always be revisited when treatment response does not yield the expected results.
Submitted: November 15, 2019; accepted February 28, 2020.
Published online: July 23, 2020.
Potential conflicts of interest: None.
Funding/support: None.
Patient consent: Consent was received from the patient to publish this case report, and information has been de-identified to protect anonymity.
REFERENCES
1. Wechsler D. WAIS-IV: Wechsler Adult Intelligence Scale. San Antonio, TX: Psychological Corporation & Pearson Education, Inc; 2008.
2. Wechsler D. Wechsler Test of Adult Reading: WTAR. San Antonio, TX: Psychological Corporation; 2001.
3. Griffiths S, Sherman EMS, Strauss E. Dementia Rating Scale-2. In: Kreutzer JS, DeLuca J, Caplan B (eds). Encyclopedia of Clinical Neuropsychology. Springer, New York, NY; 2011.
4. Patterson J. Controlled Oral Word Association Test. In: Kreutzer JS, DeLuca J, Caplan B (eds). Encyclopedia of Clinical Neuropsychology. Springer, New York, NY; 2011.
5. Reitan RM. Trail Making Test. Tucson, AZ: Reitan Neuropsychology Laboratory; 1992.
6. Berg EA, Grant DA, Heaton RK. Wisconsin Card Sorting Test. Odessa, FL: Psychological Corporation; 1993.
7. Knight JA, Kaplan E. The Handbook of Rey-Osterrieth Complex Figure Usage: Clinical and Research Applications. Lutz, FL: Psychological Assessment Resources, Inc; 2003.
8. Delis DC, Kramer JH, Kaplan E, et al. California Verbal Learning Test-Children’s Version (CVLT-C). San Antonio, TX: Psychological Corporation; 1994.
9. Randolph C. RBANS Update: Repeatable Battery for the Assessment of Neuropsychological Status. Bloomington, MN: NCS Pearson; 2012.
10. Kaplan E, Goodglass H, Weintraub S. Boston Naming Test. Philadelphia, PA: Lea & Febiger; 1983.
11. Olderbak S, Wilhelm O, Olaru G, et al. A psychometric analysis of the Reading the Mind in the Eyes Test: toward a brief form for research and applied settings. Front Psychol. 2015;6:1503. PubMed CrossRef
12. Eknoyan D, Hurley RA, Taber KH. The Clock Drawing Task: common errors and functional neuroanatomy. J Neuropsychiatry Clin Neurosci. 2012;24(3):260–265. PubMed CrossRef
13. Maeshima S, Osawa A, Maeshima E, et al. Usefulness of a Cube-Copying Test in outpatients with dementia. Brain Inj. 2004;18(9):889–898. PubMed CrossRef
14. Beck AT, Steer RA. Manual for the Beck Depression Inventory. San Antonio, TX: The Psychological Corporation; 1987.
15. Beck AT, Steer RA. Beck Anxiety Inventory Manual. San Antonio, TX: Psychological Corporation; 1993.
16. Mioshi E, Dawson K, Mitchell J, et al. The Addenbrooke’s Cognitive Examination Revised (ACE-R): a brief cognitive test battery for dementia screening. Int J Geriatr Psychiatry. 2006;21(11):1078–1085. PubMed CrossRef
17. Shetty S, Pitti V, Satish Babu CL, et al. Bruxism: a literature review. J Indian Prosthodont Soc. 2010;10(3):141–148. PubMed CrossRef
18. Petrides M. The role of the mid-dorsolateral prefrontal cortex in working memory. Exp Brain Res. 2000;133(1):44–54. PubMed CrossRef
19. Goldman-Rakic PS. Circuitry of the frontal association cortex and its relevance to dementia. Arch Gerontol Geriatr. 1987;6(3):299–309. PubMed CrossRef
20. Rolls ET. Précis of The brain and emotion. Behav Brain Sci. 2000;23(2):177–191, discussion 192–233. PubMed CrossRef
21. Leh SE, Petrides M, Strafella AP. The neural circuitry of executive functions in healthy subjects and Parkinson’s disease. Neuropsychopharmacology. 2010;35(1):70–85. PubMed CrossRef
22. Barker RA, Williams-Gray CH. Review: the spectrum of clinical features seen with alpha synuclein pathology. Neuropathol Appl Neurobiol. 2016;42(1):6–19. PubMed CrossRef
23. Kovacs GG. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol. 2015;41(1):3–23. PubMed CrossRef
24. Spillantini MG, Goedert M, Crowther RA, et al. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A. 1997;94(8):4113–4118. PubMed CrossRef
25. Taki M, Ishii K, Fukuda T, et al. Evaluation of cortical atrophy between progressive supranuclear palsy and corticobasal degeneration by hemispheric surface display of MR images. AJNR Am J Neuroradiol. 2004;25(10):1709–1714. PubMed
26. Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology. 2008;246(1):214–221. PubMed CrossRef
27. Taylor J-P, O’Brien J. Neuroimaging of dementia with Lewy bodies. Neuroimaging Clin N Am. 2012;22(1):67–81, viii. PubMed CrossRef
28. Matsusue E, Fujii S, Kanasaki Y, et al. Cerebellar lesions in multiple system atrophy: postmortem MR imaging-pathologic correlations. AJNR Am J Neuroradiol. 2009;30(9):1725–1730. PubMed CrossRef
29. Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670–676. PubMed CrossRef
30. Agarwal R, Sze G. Neuro-lyme disease: MR imaging findings. Radiology. 2009;253(1):167–173. PubMed CrossRef
31. Hildenbrand P, Craven DE, Jones R, et al. Lyme neuroborreliosis: manifestations of a rapidly emerging zoonosis. AJNR Am J Neuroradiol. 2009;30(6):1079–1087. PubMed CrossRef
32. Niemantsverdriet E, Valckx S, Bjerke M, et al. Alzheimer’s disease CSF biomarkers: clinical indications and rational use. Acta Neurol Belg. 2017;117(3):591–602. PubMed CrossRef
33. Khan BK, Yokoyama JS, Takada LT, et al. Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry. 2012;83(4):358–364. PubMed CrossRef
34. Rademakers R, Neumann M, Mackenzie IRA. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8(8):423–434. PubMed CrossRef
35. Pakrasi S, O’Brien JT. Emission tomography in dementia. Nucl Med Commun. 2005;26(3):189–196. PubMed CrossRef
36. Morbelli S, Ferrara M, Fiz F, et al. Mapping brain morphological and functional conversion patterns in predementia late-onset bvFTD. Eur J Nucl Med Mol Imaging. 2016;43(7):1337–1347. PubMed CrossRef
37. Vannini P, Hanseeuw B, Munro CE, et al. Hippocampal hypometabolism in older adults with memory complaints and increased amyloid burden. Neurology. 2017;88(18):1759–1767. PubMed CrossRef
Enjoy this premium PDF as part of your membership benefits!



