Introduction: Vilazodone, a selective and potent 5-HT1A partial agonist and 5-HT reuptake inhibitor, has been approved for treatment of major depressive disorder (MDD) in adults. The primary objective of the study was to compare the efficacy and tolerability of switching to 3 different doses of vilazodone from an equivalent dose range of generic selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs) in adult subjects with MDD.
Method: This was an 8-week, randomized, double-blind, parallel-group, 3-arm trial to compare vilazodone 10 mg/d, 20 mg/d, and 40 mg/d as starting doses. Data were collected from December 2012 to December 2013. There was no washout phase, prior medications were stopped at the baseline visit, and vilazodone was started the next day in adults with MDD (DSM-IV criteria). The 10-mg/d and 20-mg/d dose was increased to 40 mg/d by week 3 and week 1, respectively, and the 40-mg/d initiation dose continued unchanged. The primary efficacy measure was change in Montgomery-Asberg Depression Rating Scale (MADRS) score between the 3 dose groups. The secondary efficacy measures were changes in Clinical Global Impressions-Severity (CGI-S), CGI-Improvement (CGI-I), and Hamilton Anxiety Rating Scale (HARS) scores. Safety measures were obtained by spontaneously reported adverse events, vital signs recording, and laboratory tests. Multivariate tests were used for statistical analysis.
Results: Seventy subjects were randomized, and 60 subjects completed the study (n = 20 in each group). Overall, there was a significant reduction in MADRS score from baseline (26.08 ± 1.1) to week 8 (9.86 ± 1.2) in the entire sample (P < .001). Similarly, there was a significant improvement in CGI-S (P < .001), CGI-I (P < .001) and HDRS (P < .001) scores from baseline to the end of the trial. There were no significant differences between the 3 vilazodone dose-initiation groups in changes in MADRS scores (P = .95) or changes in CGI-S (P = .83), CGI-I (P = .51), or HARS scores (P = .61). Dry mouth (n = 55), nausea (n = 10), and diarrhea (n = 5) were the most common side effects, with diarrhea reported in 5 subjects in the 40-mg/d initiation group. No serious adverse events were reported.
Conclusions: The present study indicates the potential benefit of switching to vilazodone in patients with MDD who are inadequate responders to SSRIs or SNRIs. There were no meaningful differences in efficacy or tolerability between the 3 different dose-initiation strategies with vilazodone; however, diarrhea appeared to be more frequently reported with the 40-mg/d dose. Given the modest sample size, larger studies are required to confirm our findings.
Trial Registration: ClinicalTrials.gov identifiers: NCT02015546 and NCT01473381
An 8-Week Randomized, Double-Blind Trial Comparing Efficacy, Safety, and Tolerability of 3 Vilazodone Dose-Initiation Strategies Following Switch From SSRIs and SNRIs in Major Depressive Disorder

ABSTRACT
Introduction: Vilazodone, a selective and potent 5-HT1A partial agonist and 5-HT reuptake inhibitor, has been approved for treatment of major depressive disorder (MDD) in adults. The primary objective of the study was to compare the efficacy and tolerability of switching to 3 different doses of vilazodone from an equivalent dose range of generic selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs) in adult subjects with MDD.
Method: This was an 8-week, randomized, double-blind, parallel-group, 3-arm trial to compare vilazodone 10 mg/d, 20 mg/d, and 40 mg/d as starting doses. Data were collected from December 2012 to December 2013. There was no washout phase, prior medications were stopped at the baseline visit, and vilazodone was started the next day in adults with MDD (DSM-IV criteria). The 10-mg/d and 20-mg/d dose was increased to 40 mg/d by week 3 and week 1, respectively, and the 40-mg/d initiation dose continued unchanged. The primary efficacy measure was change in Montgomery-Asberg Depression Rating Scale (MADRS) score between the 3 dose groups. The secondary efficacy measures were changes in Clinical Global Impressions-Severity (CGI-S), CGI-Improvement (CGI-I), and Hamilton Anxiety Rating Scale (HARS) scores. Safety measures were obtained by spontaneously reported adverse events, vital signs recording, and laboratory tests. Multivariate tests were used for statistical analysis.
Results: Seventy subjects were randomized, and 60 subjects completed the study (n = 20 in each group). Overall, there was a significant reduction in MADRS score from baseline (26.08 ± 1.1) to week 8 (9.86 ± 1.2) in the entire sample (P < .001). Similarly, there was a significant improvement in CGI-S (P < .001), CGI-I (P < .001) and HDRS (P < .001) scores from baseline to the end of the trial. There were no significant differences between the 3 vilazodone dose-initiation groups in changes in MADRS scores (P = .95) or changes in CGI-S (P = .83), CGI-I (P = .51), or HARS scores (P = .61). Dry mouth (n = 55), nausea (n = 10), and diarrhea (n = 5) were the most common side effects, with diarrhea reported in 5 subjects in the 40-mg/d initiation group. No serious adverse events were reported.
Conclusions: The present study indicates the potential benefit of switching to vilazodone in patients with MDD who are inadequate responders to SSRIs or SNRIs. There were no meaningful differences in efficacy or tolerability between the 3 different dose-initiation strategies with vilazodone; however, diarrhea appeared to be more frequently reported with the 40-mg/d dose. Given the modest sample size, larger studies are required to confirm our findings.
Trial Registration: ClinicalTrials.gov identifiers: NCT02015546 and NCT01473381
Prim Care Companion CNS Disord 2015;17(4):doi:10.4088/PCC.14m01734
© Copyright 2015 Physicians Postgraduate Press, Inc.
Submitted: October 9, 2014; accepted March 27, 2015.
Published online: August 6, 2015.
Corresponding author: Ashwin A. Patkar, MD, Department of Psychiatry, Duke University Medical Center, 2218 Elder St, Ste 127, Durham, NC 27705 ([email protected]).
As an increasing number of antidepressants become generically available, the generic penetration of the antidepressant market in the United States has increased from about 41% in 2004 to about 73% in 2010.1 Although generic selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) are widely used, they continue to have several limitations including variability of response, intolerable side effects, and high discontinuation rates. In the real-world setting, clinicians have to frequently switch patients between different antidepressants for efficacy and tolerability reasons and need scientific data to make informed decisions regarding the switching strategies and the outcome for patients following the switch. In the current climate of managed care and formulary cost containment, such information is often mandated by insurance providers prior to authorizing a switch to a branded product.
Vilazodone, a selective and potent 5-HT1A partial agonist and reuptake inhibitor, has been approved for treatment of major depressive disorder (MDD) in adults. On the basis of its mechanism of action, vilazodone offers utility in terms of improved tolerability. One of the potential limitations of initiating vilazodone is the initial dose titration required to minimize nausea and vomiting that were reported more frequently with the drug compared to placebo.2,3 The current recommendation is to start with 10 mg/d of vilazodone and titrate to the effective therapeutic dose of 40 mg/d in 2 weeks. There are no data on whether switching to a higher dose of vilazodone may be clinically acceptable and the extent to which discontinuation symptoms from stopping a previous antidepressant influence initiation of vilazodone. Moreover, there is no information to show that patients do not suffer adverse outcomes (eg, no worsening of symptoms) or may indeed clinically benefit following a switch to vilazodone. The study objectives were as follows:
- Primary objective: To compare the safety and tolerability of switching to 3 different doses of vilazodone (10 mg/d, 20 mg/d, 40 mg/d) from an equivalent dose range of generic SSRIs or SNRIs in patients with MDD.
- To compare the efficacy of switching to 3 different doses of vilazodone (10 mg/d, 20 mg/d, 40 mg/d) from an equivalent dose range of generic SSRIs or SNRIs in patients with MDD.
- To examine the rate and extent of discontinuation syndrome following the switch from generic SSRIs and SNRIs to 3 doses of vilazodone.
- To compare the rate and extent of discontinuation symptoms following abrupt discontinuation versus a 1-week taper of vilazodone at the end of the 8-week trial.
METHOD
This was an 8-week, randomized, double-blind, parallel-group, 3-arm trial. The study sites were Duke University Medical Center, Durham, North Carolina, and Carolina Behavioral Care, Durham, North Carolina (ClinicalTrials.gov identifiers: NCT02015546 and NCT01473381). Data were collected from December 2012 to December 2013.
Patients who were switched from existing antidepressants for tolerability or efficacy reasons had their SSRI or SNRI dose reduced to fluoxetine 20 mg/d, citalopram 20 mg/d, escitalopram 10 mg/d, paroxetine 20 mg/d, sertraline 50 mg/d, or venlafaxine 75 mg/d for at least 1 week and then abruptly discontinued. Vilazodone was initiated the next day at doses of 10 mg/d, 20 mg/d, or 40 mg/d. The 10-mg/d dose was titrated to 40 mg/d as per the dose titration scheme recommended in the package insert4 (10 mg/d in week 1, 20 mg/d in week 2, 40 mg/d in week 3), the 20-mg/d dose was titrated to 40 mg/d in 1 week, and the 40-mg/d initiation dose continued unchanged. Subjects unable to tolerate the 40-mg/d dose were reduced to the 20-mg/d dose. At the end of the 8-week trial, subjects in each group were randomly assigned to an abrupt discontinuation or a 1-week taper of vilazodone to assess differences in discontinuation symptoms. A week-9 follow-up assessment of discontinuation symptoms was performed. Any new medication started during the study was evaluated by the investigator to determine its effect on study integrity and subject safety. Subjects and investigators were blinded to study drug and doses. The rating scales used were the Montgomery-Asberg Depression Rating Scale (MADRS),5-7 Hamilton Depression Rating Scale (HDRS),8,9 Hamilton Anxiety Rating Scale (HARS),10 Sheehan Disability Scale (SDS),11 Clinical Global Impressions-Severity (CGI-S), and CGI-Improvement (CGI-I).12
- This study investigated the safety and effectiveness of vilazodone (10 mg/d, 20 mg/d, 40 mg/d) in improving symptoms in patients with major depressive disorder (MDD) who are inadequate responders to selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs).
- There was a significant improvement in efficacy measures from baseline to the end of the study; safety and tolerability scores gradually decreased from baseline to the end of the study.
- Most of the treatment-emergent adverse events were from mild to moderate severity like dry mouth, nausea, weight gain, and diarrhea (only in the 40-mg dose group).
- This study indicates the potential benefit of switching to vilazodone in patients with MDD who are inadequate responders to SSRIs or SNRIs.
Inclusion Criteria
- Age 18-65 years inclusive.
- DSM-IV diagnosis of MDD.
- If female, nonpregnant/nonlactating.
- If a sexually active woman of reproductive potential, must be using adequate contraception (ie, oral contraceptives, barrier protection, or prior tubal ligation).
- Inadequate response to antidepressants: having a score ≥ 14 on the 17-item HDRS or a CGI-S score ≥ 3 after a retrospective confirmation of an adequate trial of a single antidepressant (defined as a ≥ 6-week trial of acceptable therapeutic dose [fluoxetine ≥ 40 mg, citalopram 30 mg, escitalopram 20 mg, paroxetine controlled release 37.5 mg, sertraline 150 mg, fluvoxamine 100 mg, venlafaxine extended release 225 mg]).
- Lack of tolerability of antidepressants: patient reports of side effects judged to be clinically meaningful by the investigator.
- HDRS item 2 score ≥ 2 at screening.
- Duration of current MDD ≥ 4 weeks and < 24 months.
Exclusion Criteria
- Any Axis I disorder within previous 6 months of screening except generalized anxiety disorder, social anxiety disorder, panic disorder, and simple phobias.
- MDD with postpartum onset, psychotic features, or seasonal features.
- DSM-IV substance abuse or dependence in the previous 6 months.
- Medically unstable as judged by study investigators on clinical or laboratory findings.
- Lack of capacity to provide informed written consent to investigators.
- Previous intolerance to vilazodone or current use of vilazodone at screening or within 3 months of study entry.
- Significant suicide risk as judged by the investigator based on information collected on the Columbia Suicide Severity Rating Scale.13
- History of augmentation with atypical antipsychotics, lithium, T3, or another antidepressant within 3 months of screening.
- Failure of ≥ 3 adequate trials of different antidepressants for the current episode of MDD.
Concomitant Medications
All medications for preexisting medical conditions were permitted to continue unchanged provided subjects were on a stable dose of at least 12 weeks’ duration. Subjects taking concomitant mood stabilizers or atypical antipsychotics required a 2-week washout prior to screening visit. Subjects on a minimum of 3 months of a stable dose of hypnotics (eg, zolpidem 10 mg/d or benzodiazepine dose ≤ 2 mg/d of lorazepam or trazodone ≤ 100 mg/d or quetiapine ≤ 100 mg/d) were allowed to continue their hypnotic medication at the same dose. Quetiapine at doses ≤ 100 mg/d is appropriate only for hypnotic effects. Over-the-counter medications were permitted if, in the opinion of the investigator, they were not considered to have any significant impact on the study. Any medication that, in the judgment of the investigator, had the potential to cause a clinically significant drug interaction with vilazodone required a washout.
Subjects
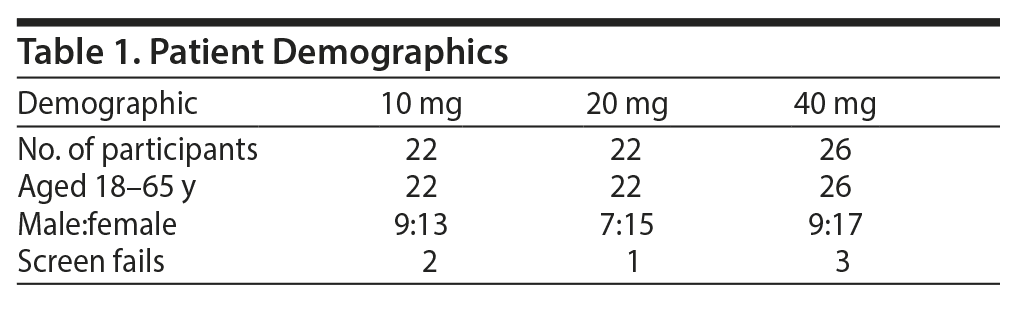
Seventy subjects were randomized; 60 subjects completed the study, and 10 were screen fails. The mean ± SD age of the subjects was 45 ± 9 years; 36 were women and 35 were men (Table 1).
Click figure to enlarge
Study Medication
Arm 1: vilazodone 10-mg/d arm (10-mg/d initiation dose, titrated to 40 mg/d in 2 weeks, continued for 8-week trial).
Arm 2: vilazodone 20-mg arm (20-mg/d initiation dose, titrated to 40 mg/d in 1 week, continued for 8-week trial).
Arm 3: vilazodone 40-mg/d arm (40-mg/d initiation and continuation dose for 8-week trial). Subjects who did not tolerate 40 mg/d had their dose reduced to 20 mg/d.
Study Visits
Study visits included screening, baseline, week 1, week 2, week 3, week 4, week 6, week 8, and week 9. Assessments were performed at each visit.
Efficacy Measures
The primary outcome measure was change in total MADRS scores from baseline to endpoint. Secondary outcomes were changes in scores on the HDRS, HARS, SDS, CGI-I, and CGI-S scores. MADRS response (defined as ≥ 50% decrease from baseline) and MADRS remission (defined as MADRS score < 10) were secondary efficacy measures.
Safety/Tolerability Measures
The primary tolerability measure for discontinuation symptoms was the Discontinuation Emergent Signs and Symptoms (DESS) checklist.14 Discontinuation symptoms that did not respond to education and supportive psychotherapy were managed by reinstituting the last dose of vilazodone at which patients did not experience discontinuation symptoms and slowly tapering the dose during 1 week or longer if necessary. Safety and tolerability were assessed by spontaneously reported adverse events, Arizona Sexual Experience Scale (ASEX) scores,15 vital signs, physical examination findings, weight and body mass index measurements, electrocardiogram and clinical laboratory assessments, and evaluation of reasons for dropout from the trial.
Data Analysis
A power analysis was conducted to provide 80% power at α = .05 to detect a difference of at least 3.0 points (SD ± 10.0, effect size = 0.30) in mean change from baseline to week 8 in MADRS total score between the baseline and end of treatment scores in each of the 3 groups. Power analysis was performed using an Internet-based software program. The results showed that a sample size of n = 60 (n = 20 in each of the 3 groups) could detect a real effect with α = .05, unless the true mean difference is quite minimal. Subjects were evaluated by 2 methods. The primary method was an intent-to-treat (ITT) analysis, including those who discontinue drug due to lack of efficacy or due to side effects. The secondary method was computer analysis. The ITT group included randomly assigned patients receiving study drug with a postbaseline efficacy assessment. The safety population comprised all patients receiving study drug with a postbaseline safety assessment. Primary efficacy analysis was conducted in the ITT population using the last-observation-carried-forward (LOCF) method. Treatment group comparisons were based on differences in least-squares mean (LSM) changes from baseline to week 8/end of treatment from an analysis of covariance (ANCOVA) model containing terms for treatment and site with baseline MADRS score included as a covariate. ANCOVA models were used to analyze change from baseline in HDRS, HARS, and CGI-I and CGI-S scores. CGI-I scores at endpoint were assessed with analysis of variance.
Click figure to enlarge
Click figure to enlarge
Click figure to enlarge
RESULTS
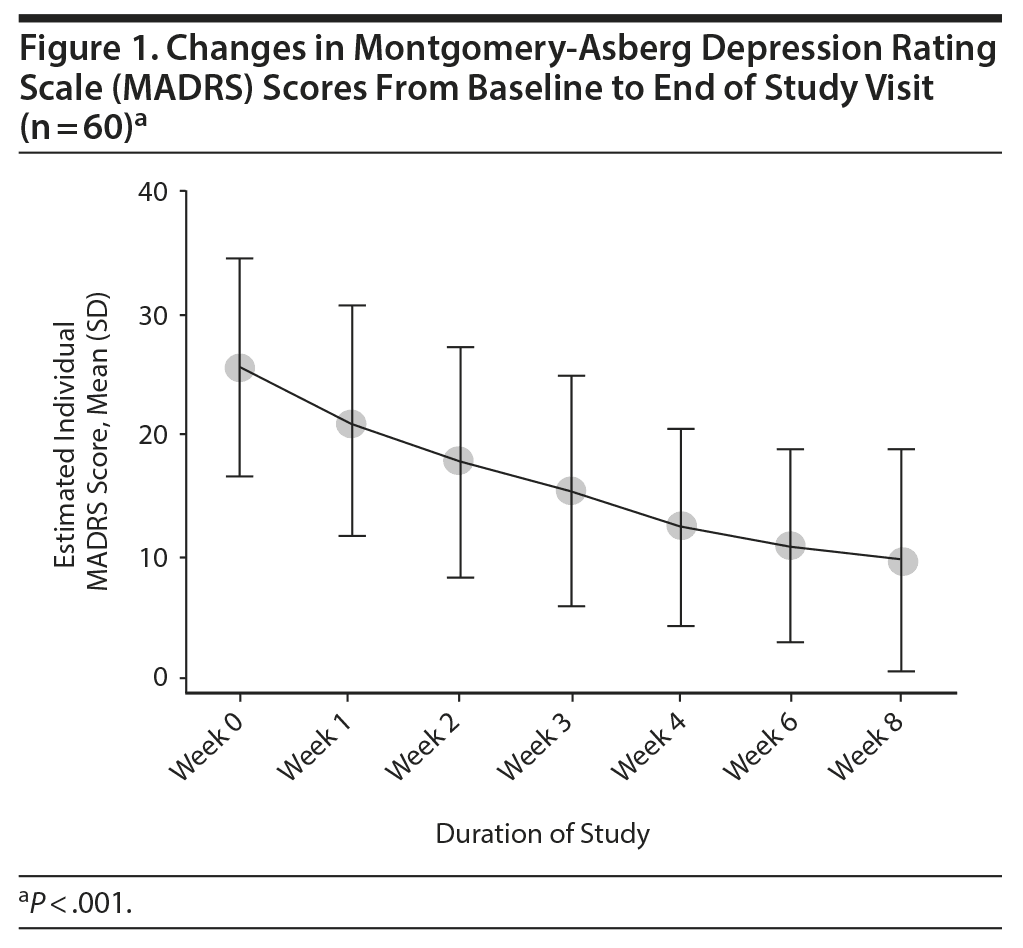
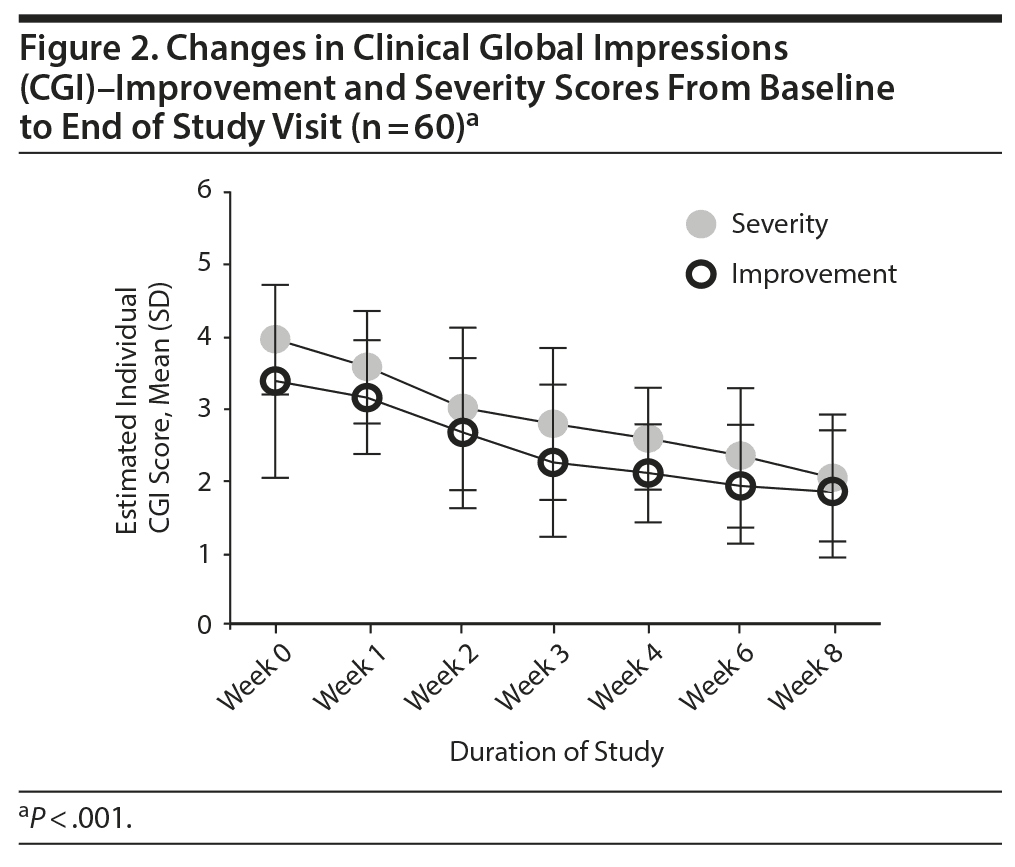
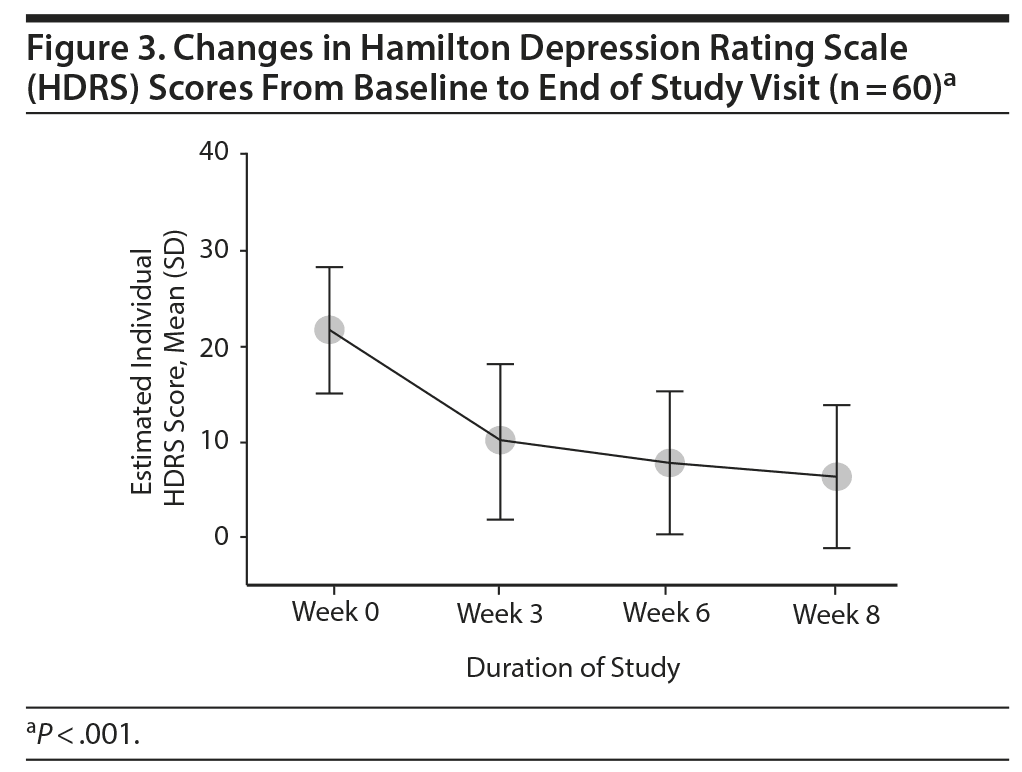
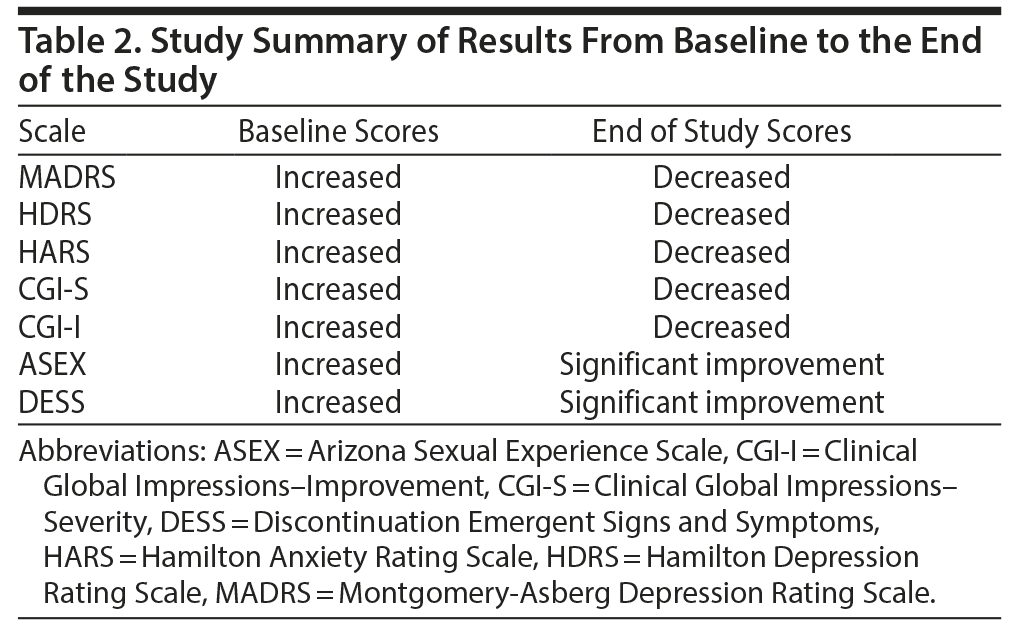
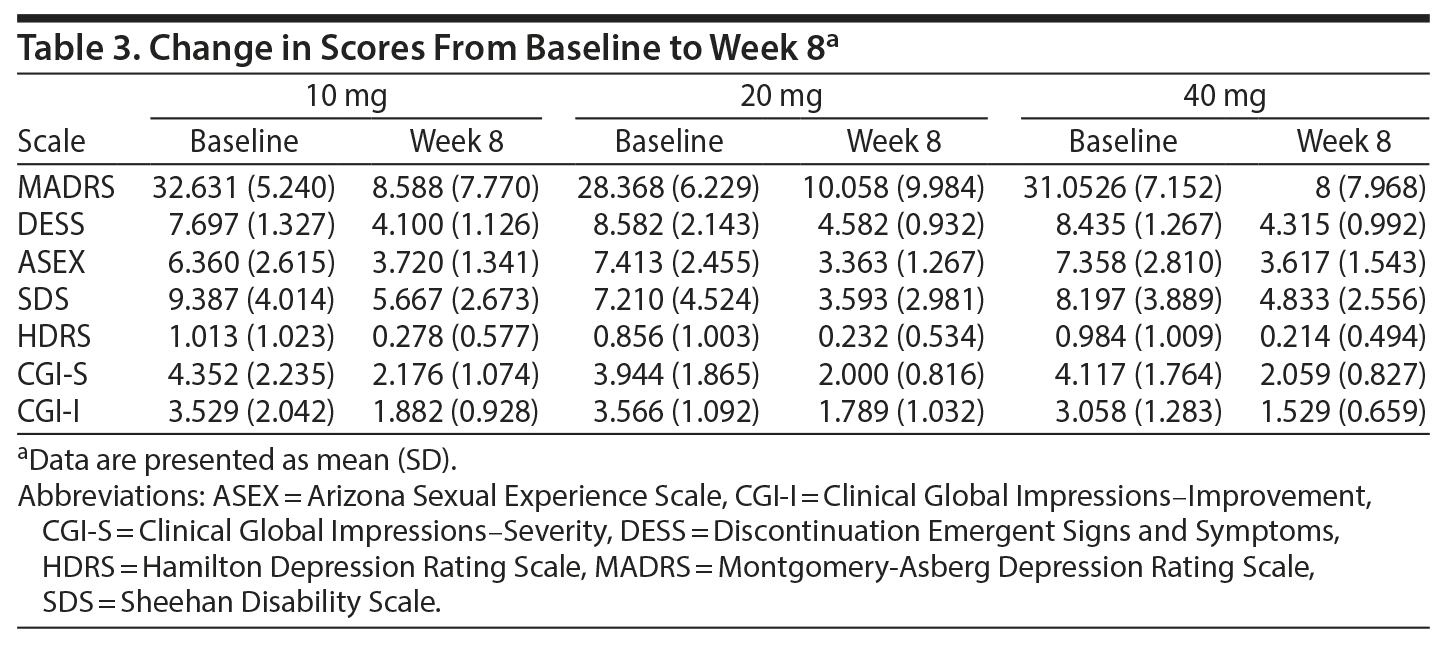
Overall, there was a significant reduction in mean ± SD MADRS score from baseline (26.08 ± 1.1) to week 8 (9.86 ± 1.2) in the entire sample (P < .001) (Figure 1). Similarly, there was a significant improvement in CGI-S scores (P < .001), CGI-I scores (P < .001) (Figure 2), and HDRS scores (P < .001) (Figure 3) from baseline to the end of the trial. There were no significant differences between the 3 vilazodone dose-initiation groups in changes in MADRS scores (P = .95) or changes in CGI-S (P = .83), CGI-I (P = .51), or HARS (P = .61) scores. SDS-associated symptoms of work (baseline: 4.8 ± 3.3, week 8: 4.10 ± 3.0, P < .01), social life (baseline: 5.20 ± 2.9, week 8: 4.4 ± 2.859, P < .01), and family and home responsibility (baseline: 4.9 ± 3.3, week 8: 4.5 ± 3.0, P < .01) showed improvement from extreme disability to mild/moderate disability. DESS scores showed a decrease from baseline (5.9 ± 9.9) to endpoint (2.2 ± 5.5, P < .01). ASEX scores showed a decrease from baseline (17.5 ± 5.2) to endpoint (15.9 ± 6.3, P = .01). There were no significant differences between the 3 vilazodone dose groups in changes in DESS score (P = .55) and ASEX scores (P = .77). Table 2 summarizes the results from baseline to the end of the study. Table 3 shows the change in scores from baseline to week 8.
Click figure to enlarge
Adverse Events
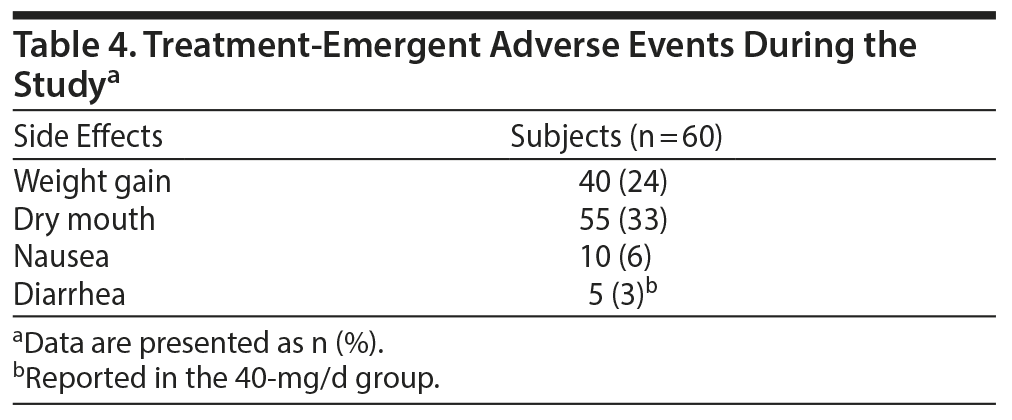
Most of the treatment-emergent adverse events were from mild to moderate severity. No patients experienced any serious adverse events. Dry mouth (n = 55), weight gain (n = 40), nausea (n = 10), and diarrhea (n = 5) were the most common side effects, with diarrhea reported in 5 subjects in the 40-mg/d initiation group. There were no serious adverse events reported. Table 4 summarizes the adverse events.
DISCUSSION
This randomized, double-blind, parallel-group study has investigated the safety and effectiveness of vilazodone in improving symptoms in patients with MDD who are inadequate responders to SSRIs or SNRIs. There was significant improvement in MADRS scores from baseline to week 8. Similarly, there was significant improvement in HDRS, HARS, CGI-I, and CGI-S scores from baseline to week 8. But the rating scales showed no significant differences between the 3 vilazodone dose-initiation groups from baseline to week 8. Effectively, SDS-associated symptoms of work, social life, and family and home responsibility showed substantial improvement from extreme disability to mild/moderate disability. As the study progressed, DESS and ASEX scores showed a gradual decrease from baseline to week 8. There were no significant differences between the 3 vilazodone dose groups in changes in DESS scores and ASEX scores.
Click figure to enlarge
Click figure to enlarge
Vilazodone provides a new option for the treatment of MDD due to its unique dual mechanism of action and may hold promise for patients who cannot tolerate or have not responded to previous antidepressant monotherapies. Furthermore, vilazodone’s use should be extended for the treatment of psychiatric conditions similar to those treated by SSRIs or SNRIs.
The present study indicates the potential benefit of switching to vilazodone in patients with MDD who are inadequate responders to SSRIs or SNRIs. There were no meaningful differences in efficacy or tolerability between the 3 different dose-initiation strategies with vilazodone; however, diarrhea appeared to be more frequently reported with the 40-mg/d dose. Given the modest sample size, larger studies are required to confirm our findings.
Drug names: citalopram (Celexa and others), escitalopram (Lexapro and others), fluoxetine (Prozac and others), fluvoxamine (Luvox and others), lithium (Lithobid and others), lorazepam (Ativan and others), paroxetine (Paxil, Pexeva, and others), quetiapine (Seroquel), sertraline (Zoloft and others), trazodone (Oleptro and others), venlafaxine (Effexor and others), vilazodone (Viibryd), zolpidem (Ambien, Edluar, and others).
Author affiliations: Duke University Medical Center, Durham, North Carolina (Drs Millet and Patkar and Ms Rele); Carolina Behavioral Care, Durham, North Carolina (Dr Millet); Kyungpook National University Hospital, Kyungpook, Korea (Dr Sungman Kim), Kyung Hee University, Seoul, Korea (Dr Paik), Dong-A University, Busan, South Korea (Dr Seonghwan Kim); and Global Medical Education, New York, New York (Dr Masand).
Potential conflicts of interest: Dr Millet has received grant/research support from Forest Laboratories. Dr Patkar has served as a consultant to Forest, Dey, Gilead, and TTK; has received grant/research support from National Institute on Alcohol Abuse and Alcoholism, Duke Endowment, FORUM, Sunovion, Titan, Pfizer, Shire, Forest, Dey, Janssen, and Lundbeck; and has served on the speakers or advisory boards of Alkermes, Dey, Pfizer, Sunovion, and Bristol-Myers Squibb. Dr Masand has served as a consultant to Forest, Lundbeck, Merck, Pfizer, and Sunovion; has received honoraria from and served on the speakers or advisory boards of Forest, GlaxoSmithKline, Merck, Pfizer, and Sunovion; and is a stock shareholder in Global Medical Education. Drs Sungman Kim, Paik, and Seonghwan Kim and Ms Rele report no conflicts of interest related to the subject of this article.
Funding/support: This study was supported by Forest Laboratories through an Investigator Initiated Award.
Role of the sponsor: The sponsor provided funding support for an investigator-initiated grant (principal investigator: Dr Patkar). The sponsor played no role in protocol design, conduct of the trial, data analysis, and interpretation of manuscript production.
REFERENCES
1. Ventimiglia J, Kalali AH. Generic penetration in the retail antidepressant market. Psychiatry (Edgmont). 2010;7(6):9-11. PubMed
2. Khan A, Cutler AJ, Kajdasz DK, et al. A randomized, double-blind, placebo-controlled, 8-week study of vilazodone, a serotonergic agent for the treatment of major depressive disorder. J Clin Psychiatry. 2011;72(4):441-447. PubMed doi:10.4088/JCP.10m06596
3. Rickels K, Athanasiou M, Robinson DS, et al. Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(3):326-333. PubMed doi:10.4088/JCP.08m04637
4. VIIBRYD [package insert]. St Louis, Missouri: Forest Laboratories, Inc; 2011.
5. Hawley CJ, Gale TM, Sivakumaran T; Hertfordshire Neuroscience Research group. Defining remission by cut off score on the MADRS: selecting the optimal value. J Affect Disord. 2002;72(2):177-184. PubMed doi:10.1016/S0165-0327(01)00451-7
6. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382-389. PubMed doi:10.1192/bjp.134.4.382
7. Zimmerman M, Posternak MA, Chelminski I. Defining remission on the Montgomery-Asberg depression rating scale. J Clin Psychiatry. 2004;65(2):163-168. PubMed doi:10.4088/JCP.v65n0204
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56-62. PubMed doi:10.1136/jnnp.23.1.56
9. Hamilton M. Rating depressive patients. J Clin Psychiatry. 1980;41(12 pt 2):21-24. PubMed
10. Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959;32(1):50-55. PubMed doi:10.1111/j.2044-8341.1959.tb00467.x
11. Sheehan KH, Sheehan DV. Assessing treatment effects in clinical trials with the discan metric of the Sheehan Disability Scale. Int Clin Psychopharmacol. 2008;23(2):70-83. PubMed doi:10.1097/YIC.0b013e3282f2b4d6
12. Guy W. Clinical Global Impressions (028-CGI). In: ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: National Institute of Mental Health; 1976:216-222.
13. Posner K, Brent D, Lucas C, et al. Columbia-Suicide Severity Rating Scale (C-SSRS). http://www.cssrs.columbia.edu/docs/ C-SSRS_1_14_09_Baseline.pdf. Accessed September 22, 2011.
14. Rosenbaum JF, Zajecka J. Clinical management of antidepressant discontinuation. J Clin Psychiatry. 1997;58(suppl 7):37-40. PubMed
15. McGahuey CA, Gelenberg AJ, Laukes CA, et al. The Arizona Sexual Experience Scale (ASEX): reliability and validity. J Sex Marital Ther. 2000;26(1):25-40. PubMed doi:10.1080/009262300278623
Enjoy this premium PDF as part of your membership benefits!


