Methylphenidate Transdermal System: A Multisite, Open-Label Study of Dermal Reactions in Pediatric Patients Diagnosed With ADHD
Objective: To characterize dermal reactions and examine methylphenidate (MPH) sensitization in subjects receiving methylphenidate transdermal system (MTS).
Method: This multicenter, open-label, dose-optimization study utilized MTS doses of 10, 15, 20, and 30 mg in children aged 6 to 12 years, inclusive (N = 305), with a DSM-IV-TR primary diagnosis of attention-deficit/hyperactivity disorder. The study was conducted between January 8, 2007, and August 23, 2007. Subjects wore MTS on their hips for 9 hours per day, alternating sides daily for a total of 7 weeks. Assessments included the Experience of Discomfort scale, Transdermal System Adherence scale, and Dermal Response Scale (DRS; 0 = no irritation, 7 = strong reaction). On-study reevaluations were conducted to characterize DRS scores ≥ 4. Epicutaneous allergy patch testing was conducted for DRS scores ≥ 6, persistent DRS scores ≥ 4, DRS score increase following an assessment of ≥ 4, or DRS scores of 4 or 5 following elective discontinuation.
Results: Approximately half of subjects experienced definite erythema at the patch site that generally dissipated within 24 hours. Four subjects experienced a DRS score of 4 (1%): erythema in 1 subject resolved on study treatment, 2 cases resolved poststudy and subjects tolerated oral MPH, and 1 subject discontinued treatment. The latter subject was referred for patch testing and was diagnosed with allergic contact sensitization to MPH.
Conclusions: Few severe dermal effects were seen with MTS treatment. Dermal reactions were characterized as contact dermatitis and dissipated rapidly. On patch testing, 1 subject (0.3%) manifested sensitization to MPH.
Trial Registration: clinicaltrials.gov Identifier: NCT00434213
Prim Care Companion J Clin Psychiatry 2010;12(6):e1-e9
© Copyright Physicians Postgraduate Press, Inc.
Submitted: March 3, 2010; accepted May 21, 2010.
Published online: December 2, 2010 (doi:10.4088/PCC.10m00996pur).
Corresponding author: Erin M. Warshaw, MD, University of Minnesota, Dept 111K, 1 Veterans Dr, Minneapolis, MN 55417 ([email protected]).
Attention-deficit/hyperactivity disorder (ADHD) is a common neurobiologic disorder that affects an estimated 4 million children aged 3 to 17 years in the United States.1 ADHD is characterized by symptoms of inattention, hyperactivity, and impulsivity.2 Prominent symptoms and dysfunction may persist into adolescence in up to 85% of individuals.3 For several decades, oral psychostimulants, such as methylphenidate (MPH), have been a first-line therapy for ADHD.4 Although the mechanism of action of MPH has not been fully elucidated, it is thought to block the uptake of norepinephrine and dopamine into the presynaptic neuron and increase the release of these monoamines into the extraneuronal space in regions of the brain associated with ADHD.5,6 Because of the short half-life of MPH (2 to 3 hours), research has focused on modifying its delivery to extend the duration of action. Since the early 1980s, formulations of MPH have evolved from immediate-release tablets that require dosing 2 to 3 times daily to long-acting formulations that allow for once-daily dosing and continuous efficacy without peaks and troughs for 8 to 12 hours.4,7
In April 2006, the US Food and Drug Administration approved a methylphenidate transdermal system (MTS) form of delivery for children with ADHD aged 6 to 12 years. Transdermal delivery of MPH is a treatment option for children with ADHD who have difficulty tolerating or swallowing oral medications, or who would benefit from tailoring the duration of effect.8 The efficacy of MTS at doses of 10 mg to 30 mg, in patch sizes ranging from 12.5 cm2 to 37.5 cm2, and a wear time of 9 hours was demonstrated in 2 randomized, double-blind, placebo-controlled studies of children with ADHD.9,10 In addition, a recent study has shown that the therapeutic effects of MTS persisted for between 2 and 4 hours after patch removal, further extending the duration of ADHD symptom control.8
Skin irritation at patch sites is common with transdermal delivery systems, and sensitization to the pharmacologic agent (eg, clonidine) has been reported with some patches.11 For instance, up to 26% of subjects in previous clinical trials with MTS have been observed to manifest at least moderate skin irritation,8-10,12-15 although characterization of the skin irritation and the extent of potential sensitization remains unclear. While allergic contact sensitization associated with MTS has been reported in postmarketing reports,16 previous reports of allergic contact sensitization were not evaluated with epicutaneous patch testing; therefore, allergic contact sensitization was never confirmed. This study characterizes dermal reactions in children with ADHD treated with MTS and determines the rate of potential allergic contact sensitization to MPH from a transdermal delivery system in the clinical setting. Secondarily, the safety and efficacy of MTS are evaluated. Based on results from studies of various transdermal delivery systems,11 it was estimated that as many as 97% of subjects could manifest skin irritation, and it was hypothesized that ≤ 1% of subjects would experience sensitization to MPH as defined by subsequent patch testing.
Clinical Points
- Irritant contact dermatitis is the most likely diagnosis associated with dermal reactions in subjects receiving methylphenidate transdermal system.
- These aggregate findings suggest that mild-to-moderate erythema without pain or pruritus can be managed by alternating application sites.
- The rate of allergic contact sensitization to methylphenidate in this study was 0.3%, which was well within the expected ≤ 1%.
METHOD
Participants
Eligible subjects included boys and girls aged 6 to 12 years, inclusive, who met Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR)2 criteria for a primary diagnosis of ADHD, based on a detailed clinical evaluation. At baseline, eligible subjects had a total score ≥ 26 on the ADHD Rating Scale-Version IV (ADHD-RS-IV)17 and no comorbid illnesses that could affect safety or tolerability or interfere with the subject’s participation in the study. At screening and baseline, eligible female subjects of childbearing potential had a negative result on a urine pregnancy test, and all included subjects’ blood pressure measurements were within the 95th percentile for age, gender, and height.
Potential subjects were excluded if they met any of the following criteria: comorbid psychiatric diagnosis (except oppositional defiant disorder); risk for suicidal or violent behavior; history of suicide attempt; structural cardiac abnormality, cardiomyopathy, cardiac rhythm abnormality, or other serious cardiac problem; history of nonresponse to psychostimulants; history of seizures during the previous 2 years (except infantile febrile seizures); tic disorder; current diagnosis or family history of Gilles de la Tourette’s syndrome; conduct disorder; history of substance abuse or dependence; abnormal thyroid function; concurrent illness; treatment with hepatic and/or cytochrome P450 enzyme-altering agents; concomitant medications with central nervous system effects; skin disease, history of chronic skin disease, or sensitive skin syndrome (defined as subjects who often develop nonspecific skin irritancy reactions to bland materials); or clinical signs or symptoms of skin irritation.
Parents or legal guardians provided written informed consent, and subjects provided assent before participating in the study. An institutional review board at each study site approved all study documentation, including the protocol and the informed consent and assent documents.
Study Design

Click figure to enlarge
This was a multicenter, open-label, dose-optimization study that utilized MTS doses of 10, 15, 20, and 30 mg in children with ADHD. The study was conducted between January 8, 2007, and August 23, 2007. Subjects wore MTS on their hips for 9 hours per day, alternating sides daily. If applicable, a washout period of up to 1 week preceded initiation of study medication. Subjects then underwent stepwise titration to an acceptable dose condition during the dose-optimization period, which transpired over 4 weeks to allow titration to the highest dose, if indicated. Following the 4-week dose-optimization period, subjects were maintained on their optimized dose for 3 weeks, for a total of 7 weeks of MTS administration. A follow-up visit was scheduled approximately 30 days after the last dose of study medication to assess safety and dermal responses (Figure 1).
Dose optimization. All subjects began treatment with the 10-mg MTS dose. Treatment response, evaluated weekly, was based on ADHD-RS-IV scores, identification of adverse events (AEs), and the clinical judgment of the investigator. Treatment responses were categorized by the investigator into 1 of 3 conditions with associated actions: (1) “intolerable condition,” defined as an unacceptable safety profile that required tapering to a lower dose, if possible, or discontinuation if not; (2) “ineffective condition,” defined as a < 25% change in ADHD-RS-IV score with an acceptable safety profile that required an increase to the next patch size followed by weekly evaluation; and (3) “acceptable condition,” defined as a reduction (≥ 25%) in ADHD symptoms from baseline with minimal adverse effects. Subjects were eligible for an increase to the next dose/patch size after a minimum of 7 days (± 3 days) on the previous dose based on the overall response of the subject. Subjects with an acceptable condition could be titrated to the next patch size if the current dose was well tolerated and, in the investigator’s opinion, the subject would have received further symptom reduction with an increased MTS dose. The dose could be down-titrated once per subject during the study.
Assessments
Dermal evaluations. Visual dermal evaluations (ie, Dermal Response Scale [DRS], Experience of Discomfort [EOD] scale, Transdermal System Adherence [TSA] scale) were performed by trained study staff as described in previous MTS trials.8-10,12-15 The same evaluator examined each subject at each study visit. Dermal evaluations were conducted sequentially, beginning with the routine scheduled assessments. On-study reevaluations and poststudy evaluations were performed for subjects with dermal AEs.
Scheduled evaluations. The DRS was used to assess current and prior (previous day’s ) application sites for the presence or absence of primary skin reactions and other signs of skin irritation. Findings of erythema, edema, papules, and vesicles were graded on a 7-point scale, for which 0 = no irritation and 7 = strong reaction spreading beyond the test site. Additional dermal assessments were conducted when scores were 4 (definite edema) or greater. Study discontinuation was mandatory for subjects with a score of 6 (vesicular eruption) or greater, or if a score of ≥ 4 did not improve to ≤ 3 within 2 weeks.
The overall level of subject discomfort was assessed using the EOD scale. Scores ranged from 0 (no discomfort) to 3 (severe, intolerable discomfort). Discomfort levels of mild, moderate, or severe were attributed to symptoms of itching, burning, or other.
The TSA scale was used to estimate the percentage of the MTS surface that remained adhered to the skin. The scale ranged from 0 (≥ 90% adhered) to 4 (100% detached).
On-study reevaluations. Dermatologic reevaluations (DRS, EOD, TSA) of up to 4 additional visits over a period that was not to exceed 2 weeks were conducted to characterize the evolution of dermal responses for subjects experiencing a DRS score ≥ 4 (indicating the presence of definite edema [DRS score = 4]; erythema, edema, and papules [DRS score = 5]; vesicular eruption [DRS score = 6]; or a strong dermal reaction beyond the patch application site [DRS score = 7]).
Poststudy evaluations: allergic contact sensitization to MPH. Study-specific dermatologists, all specialists in allergic contact dermatitis, were selected to evaluate subjects in the event of suspected allergic contact sensitization, with the plan to have these dermatologists follow up with subjects who were sensitized to MPH.
Allergic contact sensitization to MPH was evaluated by epicutaneous patch testing18 in subjects who met any of the following: DRS score ≥ 6, persistent DRS score ≥ 4, increased DRS score after an assessment ≥ 4, or DRS score of 4 or 5 in subjects who were electively discontinued by the investigator. Subjects who met any 1 of these criteria were discontinued from the study and referred for 1 or more poststudy dermal evaluations, including assessment and epicutaneous patch testing, medical supervision for initial dose of oral MPH, follow-up for an unexplained rash during the first 4 weeks of oral MPH treatment, and quarterly follow-up phone calls for 1 year after discontinuation of treatment. Patch test results were interpreted according to North American Contact Dermatitis Group guidelines.19
Safety. Safety was assessed at each clinic visit (baseline, at weeks 1 through 5, week 7, and week 11) by evaluating AEs reported spontaneously, analyzing changes in vital signs, and conducting physical examinations. Dermal reactions were classified as an AE when pharmacologic treatment for the reaction was required.
Effectiveness. Change from baseline scores derived from the clinician-completed ADHD-RS-IV performed at weeks 1 through 5 and week 7 was analyzed to assess effectiveness in this study. The ADHD-RS-IV consists of 18 items that reflect the symptomatology of ADHD based on DSM-IV-TR criteria. Each item is scored from 0 (“no symptoms”) to 3 (“severe symptoms”).17 Total scale scores of all 18 items range from 0 to 54.
The Clinical Global Impressions-Severity of Illness and -Improvement scales (CGI-S and CGI-I) assess the investigator’s impression of the severity of the subject’s illness and the subject’s clinical improvement over time.20 The CGI-S was conducted at baseline using scores of 1 (“no symptoms”), 2 (“borderline symptoms”), 3 (“mild symptoms”), 4 (“moderate symptoms”), 5 (“marked symptoms”), 6 (“severe symptoms”), and 7 (“extreme symptoms”). Improvement relative to the CGI-S score was determined using the CGI-I. The CGI-I was completed at weeks 1 through 7 using scores of 1 (“very much improved”), 2 (“much improved”), 3 (“minimally improved”), 4 (“no change”), 5 (“minimally worse”), 6 (“much worse”), and 7 (“very much worse”). Results were dichotomized as either “improved” (including “very much improved” and “much improved”) or “not improved” (all other categories from “minimally improved” to “very much worse”).
Data Analysis
Based on the hypothesis that ≤ 1% of subjects risk becoming sensitized to MPH, a sample size of 300 subjects was planned, providing a 95% probability of detecting allergic contact sensitization to MTS. Dermal reactions were summarized by descriptive statistics for each MTS patch size and the overall population. The number and percentage of subjects were tabulated by incidence of dermal response levels (ie, “mild,” “moderate,” “severe”).
The paired t test was used to evaluate the mean changes in the ADHD-RS-IV score from baseline at weeks 1 through 4, 7, and 11. The CGI scores, treatment-emergent AEs (TEAEs), vital sign data, and physical examination findings were summarized using descriptive statistics. Continuous variables and categorical values were also summarized using descriptive statistics.
Click figure to enlarge
Click figure to enlarge
RESULTS
Subject Disposition
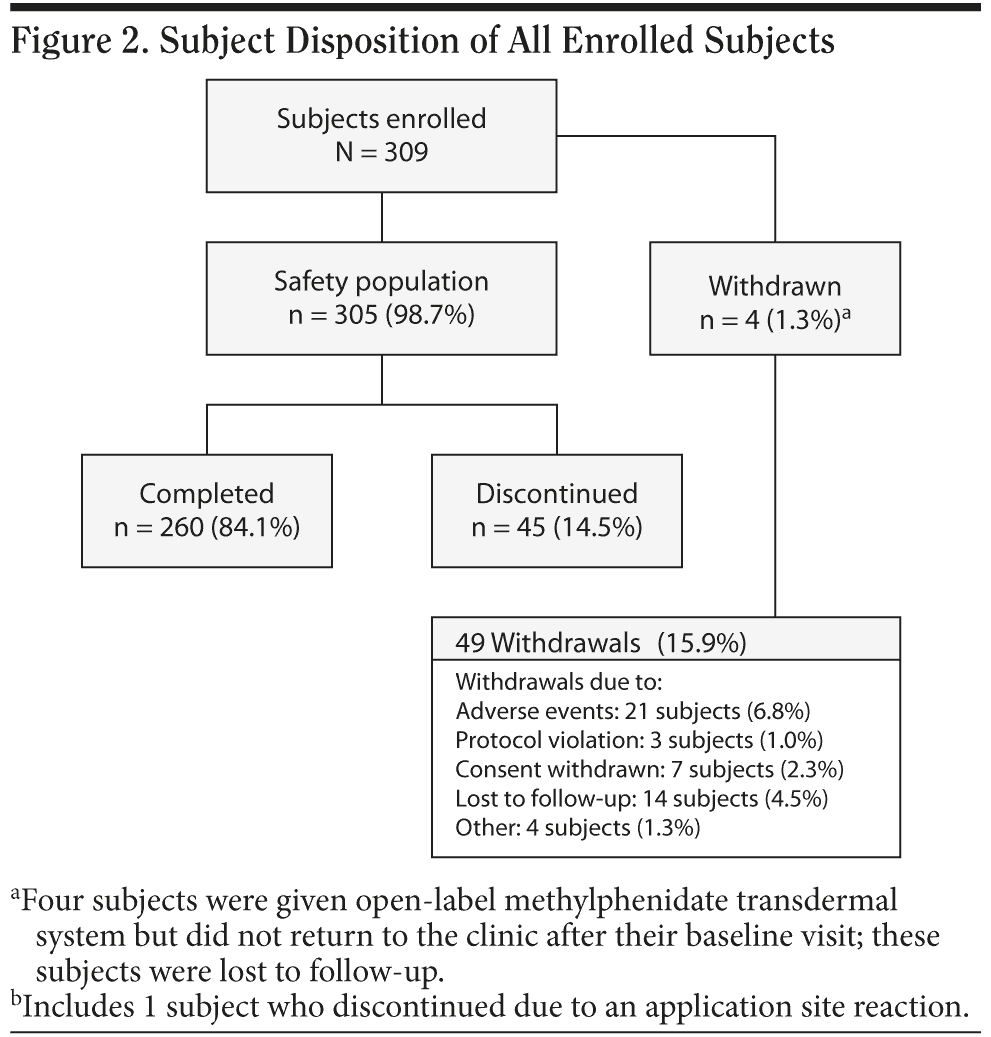
Three hundred nine subjects were enrolled, 305 subjects received at least 1 dose of study medication (safety population), 303 subjects received at least 1 dose of study medication and had both a baseline and a postbaseline measurement (intent-to-treat [ITT] population), and 260 (84.1%) completed the study (Figure 2). Of the 49 (15.9%) subject withdrawals that occurred, most were due to TEAEs or application site reactions (7.2%) or subjects who were lost to follow-up (4.5%).
Demographic and Baseline Characteristics
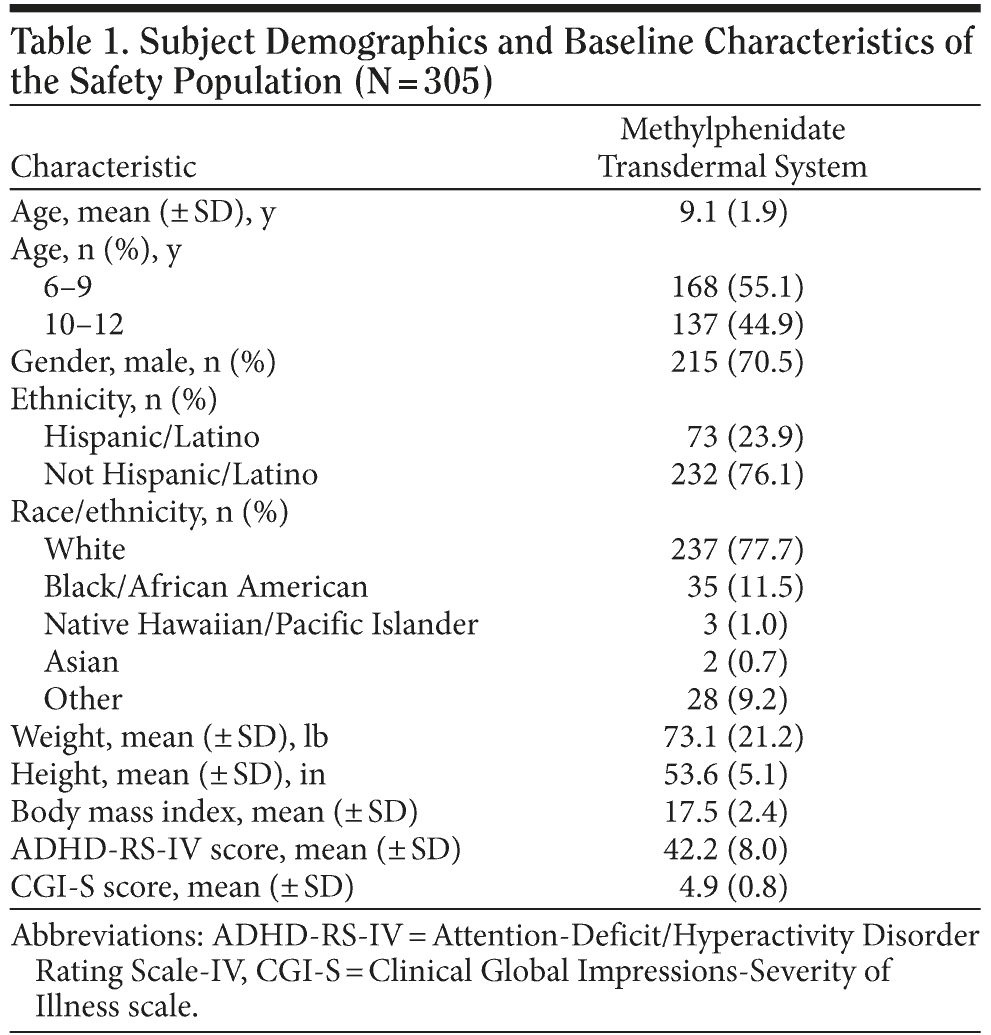
The safety population was made up of 305 subjects (Table 1). The mean subject age was 9.1 years. The majority of subjects were aged 6 to 9 years (55.1%), male (70.5%), and white (77.7%). The mean baseline ADHD-RS-IV score was 42.2 and the mean baseline CGI-S score was 4.9. The majority of subjects (62.3%) had taken or were taking ADHD medications at screening, predominantly MPH and amphetamine formulations. At baseline, all subjects had healthy, intact skin at the MTS application site; minor abnormalities (1 mole; 1 small, nearly healed abrasion; and 1 instance of dry skin) were reported in 3 subjects.
At the end of the treatment period (visit 8), 34 subjects were receiving MTS 10 mg, 84 were receiving MTS 15 mg, 70 were receiving MTS 20 mg, and 78 were receiving MTS 30 mg. One subject titrated down from 20 mg to 15 mg 2 days after visit 8 and is represented in both dose groups. Overall, the mean (± SD) MTS exposure was 45.7 (10.6) days. Specifically, for the 10-, 15-, 20-, and 30-mg MTS dose groups, the mean (± SD) exposures were 13.5 (13.2) days, 17.9 (14.5) days, 17.2 (11.5) days, and 23.4 (8.0) days, respectively.
Dermal Evaluations
Scheduled dermal assessments.
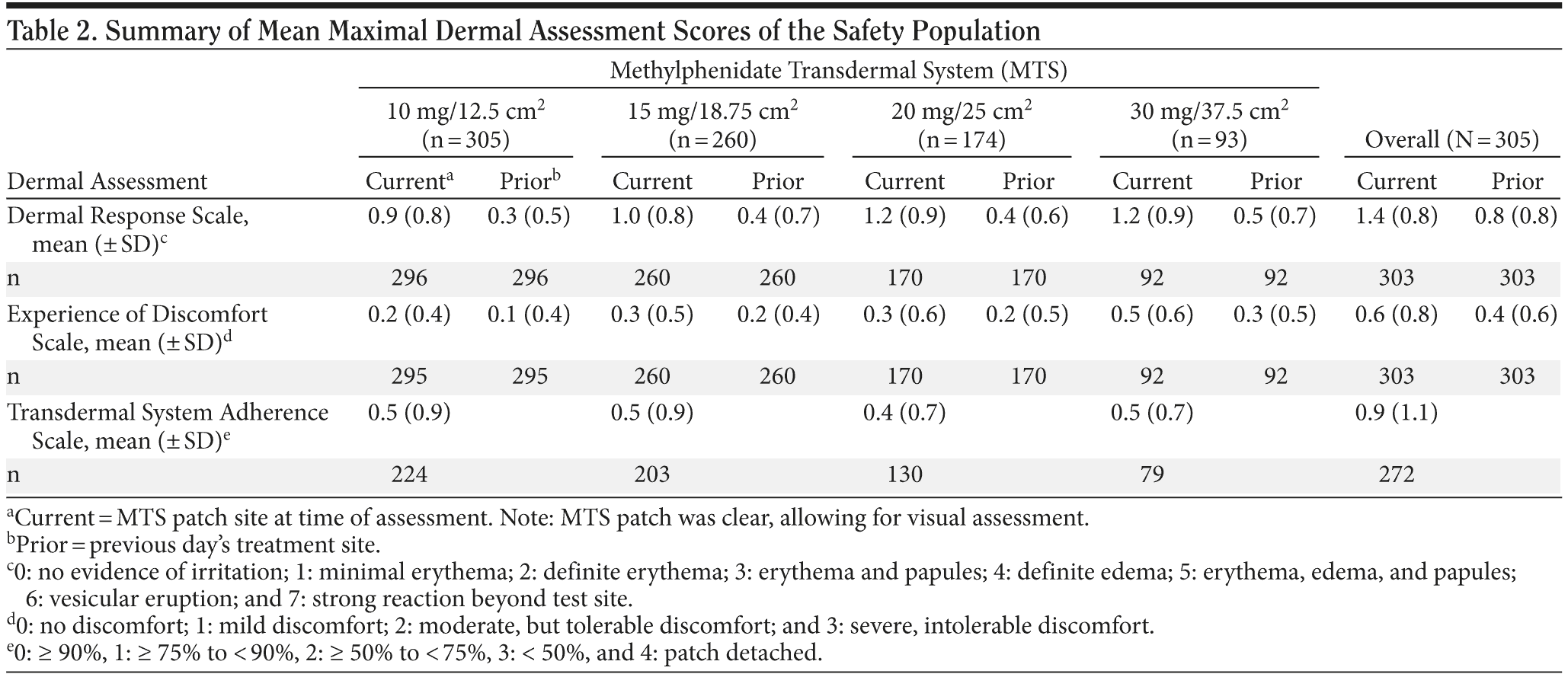
Dermal Response Scale. Evaluation of the maximal observed DRS scores measured at the current patch application site showed 15.1% of subjects experienced no dermal response (DRS score = 0), 31.8% of subjects had minimal erythema (DRS score = 1), 49.5% of subjects had definite erythema (DRS score = 2), 2.0% had erythema and papules (DRS score = 3), and 1.0% had definite edema (DRS score = 4). No subject had a DRS score of > 4 throughout the study. The DRS scores measured at the prior application site were comparatively lower than those measured at the current patch application site across all study visits. Application sites assessed on the day following patch application and removal showed that 82% of subjects had no dermal response (41%) or minimal erythema (41%).
Mean (± SD) maximal DRS scores at the current patch application site were 0.9 (0.78), 1.0 (0.85), 1.2 (0.86), and 1.2 (0.86) for the 10-mg, 15-mg, 20-mg, and 30-mg MTS doses, respectively, indicating an upward trend in the mean (± SD) maximal scores with increasing doses and patch sizes (Table 2). However, across all patch sizes the mean (± SD) maximal DRS score indicated minimal erythema at the patch site. No upward trend in mean (± SD) maximal DRS score was noted with increasing MTS patch sizes at prior patch application sites.
Click figure to enlarge
Experience of Discomfort scale. Maximal EOD score analysis revealed that > 90% of subjects experienced no discomfort (49.8%) or mild discomfort (40.7%) at the current patch application site (Table 2). At the prior application site, 64.6% and 28.5% experienced no or mild discomfort, respectively. Moderate discomfort at the current and prior application sites was reported in 8.2% and 5.9% of subjects, respectively. Less than 1% of subjects reported severe discomfort. When discomfort was reported at either the current or prior application sites, the majority of subjects reported experiencing itching.
Transdermal System Adherence scale. Overall, the mean (± SD) maximal TSA score was 0.9 (1.1), indicating ≥ 75% to < 90% of the patch was attached (Table 2). The mean TSA score ranged from 0.3 to 0.4 across all study visits, indicating successful system adherence on average. Patch detachment rates were highest during weeks 1 (2.0%) and 2 (1.7%) and declined during weeks 5 (1.0%) and 7 (0.8%).
On-study dermal reevaluations. Per protocol, additional on-study dermal reevaluations were conducted for the 4 subjects with a DRS score = 4; no subject had a DRS score > 4. Two subjects receiving MTS 15 mg or 30 mg were treated with topical corticosteroids after 4 to 7 weeks of MTS treatment. Symptoms resolved within 1 to 3 days, and MTS treatment continued without recurrence of skin symptoms. At the week 11 follow-up visit, a third subject receiving MTS had bilateral DRS scores = 4. The subject was switched to oral MPH without further incident, and skin symptoms resolved in < 7 days. The final incident of a DRS score = 4 occurred in a subject receiving MTS 15 mg. The investigator electively discontinued this subject from the study. This was the only subject who met the protocol-specified criteria for patch testing for sensitization to MPH.
Poststudy dermal evaluation for sensitization to MPH. The poststudy dermal evaluation of the electively discontinued subject with a DRS score = 4 revealed a mild reaction to MPH 10% and 0.1% in petroleum, as well as MPH 10% and 1% aqueous, which was indicative of allergic contact sensitization. The subject was instructed not to use MPH without strict medical supervision and received follow-up phone calls quarterly for 1 year. No rechallenge with oral MPH was performed and stimulant therapy continued with amphetamine. Given that this was the only subject who developed allergic contact sensitization to MPH, the rate of MPH sensitization in this study was 0.3% (1/305).
Safety Assessments
Adverse events. Overall, 743 AEs were reported in 242 (79.3%) subjects during the study period. All AEs were classified as TEAEs. The majority (> 99.5%) of TEAEs were mild or moderate in intensity. Two severe TEAEs (headache and decreased appetite) in 2 subjects (0.7%) were considered treatment related. No serious TEAEs or deaths were reported.
Twenty-two subjects (7.2%) discontinued the study because of a TEAE or an application site reaction. Reasons for discontinuation (which may have been reported in more than 1 subject) were lack of efficacy in 9 subjects, affect lability in 3 subjects, flat affect in 2 subjects, and aggression, dysphoria, irritability, tic, decreased appetite, polydipsia, muscle spasms, pollakiuria, application site discomfort, application site pain, and urticaria on thighs and abdomen in 1 subject each.
Of the 743 TEAEs, 420 events in 61.0% of subjects were considered related to study treatment. The most common (≥ 2%) TEAEs related to study treatment were decreased appetite (29.8%), insomnia (9.8%), headache (8.9%), affect lability (6.9%), irritability (5.9%), nausea (5.2%), application site pruritus (4.6%), initial insomnia (4.3%), anorexia (3.6%), decreased weight (3.3%), upper abdominal pain (3.3%), lack of efficacy (2.6%), and application site reactions (2.3%).
Adverse application site reactions. Eighteen subjects experienced application site reactions in which topical hydrocortisone or triamcinolone acetonide was administered; 13 subjects (72%) had unspecified patch reaction or itching; the remaining subjects had irritation, dermatitis, or itching/dermatitis. The duration of topical treatment ranged from 1 day to nearly 7 weeks. The majority of subjects were treated for a DRS score of 2 through 4. Three of the 4 subjects with a DRS score of 4 were treated with topical corticosteroids and reported reductions in DRS scores at the first follow-up visit. No subject required treatment with systemic corticosteroids.
Three subjects receiving MTS 10 mg or 15 mg discontinued MTS as a result of an application site reaction. No association was noted between dose and discontinuation. Two of the subjects experienced moderate discomfort or pain and itching 4 to 5 days after MTS initiation that spontaneously resolved 2 to 3 days after discontinuation. The third subject discontinued MTS at week 7 at the discretion of the investigator due to definite edema (DRS score = 4), moderate itching and burning at both current and prior patch application sites, and urticaria on the abdomen and thighs. The subject received twice-daily treatment with topical hydrocortisone 1% cream and oral diphenhydramine hydrochloride 25 mg. Within 48 hours, the subject’s symptoms resolved to minimal and definite erythema (DRS scores of 1 and 2 on prior and current sites, respectively). Urticarial lesions resolved about 8 days after MTS discontinuation. The investigator judged the urticaria to be related to treatment with MTS.
Vital signs and physical examinations. The majority of subjects had no clinically significant changes from baseline in vital signs or physical examinations. Overall mean (± SD) change from baseline to endpoint in pulse values was 3.2 (12.91) beats per minute. Changes in diastolic blood pressure from baseline to endpoint were small, showing a mean (± SD) increase from baseline of 1.8 mm Hg (8.24). Changes in systolic blood pressure from baseline to endpoint overall were similarly modest with a mean (± SD) increase from baseline of 1.1 mm Hg (10.34). No subject had diastolic blood pressure or systolic blood pressure values above the normal range at endpoint. Physical examinations for the largest proportion (88%) of subjects were within normal limits.
Effectiveness Assessments
ADHD Rating Scale-Version IV. Improvements from baseline in mean ADHD-RS-IV total scores were consistent and statistically significant from weeks 1 through 7. Overall mean (± SD) improvements (ie, decreased scores from baseline) ranged from −11.0 (10.40; P < .0001) at week 1 (the first postbaseline assessment) to -26.2 (11.48; P < .0001) at endpoint.
Clinical Global Impression-Improvement. Improvement from baseline to endpoint was observed in 86.4% of subjects on the basis of CGI-I scores. The overall proportion of subjects rated as improved rose steadily from 37.3% at the first postbaseline assessment (week 1) to 93.2% at week 8.
DISCUSSION
In the current study, the majority of subjects across MTS dose groups experienced either no dermal irritation or irritant contact dermatitis, which was limited to erythema without discernible evidence of edema or papules. In addition, most subjects experienced minimal discomfort during the study. The greater proportion of subjects had minimal or no erythema at the prior patch site compared with the current patch site, indicating that while erythema may be common with MTS, it diminishes rapidly over time. These aggregate findings suggest that mild-to-moderate erythema without pain or pruritus can be managed by alternating application sites. Findings from previous, controlled MTS studies were generally similar,8-10 illustrating that irritant contact dermatitis is the most likely diagnosis associated with dermal reactions in subjects receiving MTS.
Definite edema (DRS score = 4), the most severe reaction observed in this study, developed after several weeks of treatment in 4 cases. Treatment with topical corticosteroids is suggested when erythema does not spontaneously resolve, although the potential AEs associated with topical corticosteroid use during MTS treatment are unknown.16 In the small number of subjects who were treated with topical corticosteroids for application site reactions in the current study, all cases resolved in a timely manner, and no subject required systemic corticosteroids for a dermal reaction.
There are 3 types of dermal reactions possible with any transdermal delivery system: irritant contact dermatitis, allergic contact dermatitis, and allergic contact urticaria.21 Irritant contact dermatitis is the most common of these and has the least serious consequences. Irritant contact dermatitis is a nonimmunologic response to skin injury that generally resolves spontaneously over the course of several days after the irritant has been removed. It is associated with the development of red patches within the boundary of the patch site, mild itching, burning, and pain. Allergic contact dermatitis is a T cell-mediated, delayed hypersensitivity reaction characterized by edema or vesicles (often extending beyond the patch site boundaries) with intense itching, and sometimes burning and pain that often “crescendos” or worsens, after removal of the allergen. It is diagnosed by epicutaneous patch testing over 48 to 96 hours and can persist for several weeks. Allergic contact urticaria, the most serious and least common reaction, is a type I immunoglobulin E-mediated inflammatory response to an allergen, causing pruritus, hives, angioedema, and, in some cases, anaphylaxis within 15 minutes of exposure to the antigen. It is most commonly confirmed by skin prick testing, whereby a positive reaction, or wheal, occurs within 15 minutes.
Before the current study, the rate of allergic contact sensitization to MPH associated with MTS had not been systematically studied. At the time of this study, no cases of MPH allergic contact sensitization had been reported in clinical trials of children treated with MTS. Because it is difficult to distinguish between irritant and allergic contact dermatitis based on clinical grounds alone, criteria were developed for follow-up observation and patch testing for any subject with erythema and an indication of a more serious skin reaction. This allowed for a more accurate estimation of the risk of MPH allergic contact sensitization in a clinical setting. The hypothesis was that ≤ 1% of MTS-treated subjects would develop a dermal reaction leading to treatment discontinuation and would subsequently test positive for allergic contact dermatitis by confirmatory epicutaneous patch testing. Of the 4 children who experienced definite edema during the study, only 1 met the criteria for patch testing and was subsequently found to be sensitized to MPH. Therefore, the rate of allergic contact sensitization to MPH in this study was 0.3%, which was well within the expected ≤ 1%. The subject elected not to undergo a rechallenge with oral MPH but continued treatment with oral amphetamine without any systemic reaction.
To date, the authors are not aware of any reports in the literature regarding confirmed MTS allergic contact sensitization with follow-up oral MPH challenge definitively leading to systemic MPH sensitivity. However, data based on studies of transdermal clonidine indicate that approximately 1%-2% of individuals with confirmed allergic contact sensitization to a transdermal therapeutic system will be unable to tolerate subsequent oral administration of the therapeutic agent.22,23 Thus, given the low incidence (0.3%) of allergic contact sensitization to MTS observed in this study and the low incidence (1%-2%) of allergic contact sensitization leading to intolerability of oral administration reported in the literature, it is likely that the incidence of patients with allergic contact sensitization to MTS with subsequent intolerability to oral MPH administration would be very rare (approximately 0.003%-0.006% or 1 in 15,000 to 1 in 30,000). This estimate compares favorably with the rare occurrence of allergy to orally administered MPH reported in the literature.24
Regarding other safety parameters, MTS was associated with few serious or severe TEAEs in the present study. The most common TEAEs related to study treatment were decreased appetite, insomnia, and headache. The majority of TEAEs in the present study were mild or moderate in intensity, transient, and resolved with continued dosing. These TEAEs were similar to those reported in previous studies of MTS,8-10,12-15 as well as in studies of oral extended-release MPH in pediatric patients with the exception of dermal reactions.25-29
The effectiveness of MTS was secondarily evaluated in the current study. The majority of subjects experienced consistent improvement in ADHD symptoms over the treatment period. At the end of the treatment period, clinicians employed the 15-, 20-, and 30-mg doses equally. At the endpoint, overall scores for the ADHD-RS-IV were significantly improved (P < .0001), and 86.4% of subjects showed improvement in CGI-I scores relative to baseline CGI-S scores. These data are similar to those reported in previous studies of MTS in children with ADHD.8,9,12,13 In both laboratory- and naturalistic-setting studies, statistically significant improvements in ADHD symptoms with MTS compared with placebo have been observed in all measures and at all doses.8-13
Limitations
There were a number of methodological limitations of the current study. Conclusions about the long-term dermal effects of MTS are limited by the relatively short 14-week duration of the study. While study investigators administering and assessing MTS use at scheduled visits underwent training to detect and code dermal reactions, the intrareliability and interreliability of their DRS, EOD, and TSA ratings were not examined. Also, the spontaneous reporting of AEs employed in this study is known to underestimate the type and frequency of AEs reported over the study duration when compared to use of a stimulant-specific structured interview. Lastly, because of the nature of uncontrolled, open-label safety studies, effectiveness evaluations are limited due to potential observer bias. Nonetheless, the effectiveness results of the current study are similar to those reported in previous well-controlled efficacy studies.10,11
CONCLUSION
The results of this study indicate that dermal reactions with MTS use were predominantly mild to moderate. Dermal reactions appeared to be of an irritant contact dermatitis form; they dissipated rapidly with time and most resolved with continued treatment. Overall, less than 1% of subjects manifested sensitization to MPH.
Drug names: clonidine (Iopidine, Clorpres, and others), diphenhydramine (Benadryl and others), hydrocortisone (Acetasol, Vosol, and others), triamcinolone (Mykacet, Azmacort, and others).
Author affiliations: The University of Minnesota, Minneapolis (Dr Warshaw); Shire Development, Inc, Wayne, Pennsylvania (Drs Squires, Li, and Civil); and Northwestern University Feinberg School of Medicine, Chicago, Illinois (Dr Paller).
Potential conflicts of interest: Dr Warshaw has served as a consultant to Shire. Dr Squires is a full-time employee of Shire and a stock shareholder in Johnson & Johnson, Pfizer, and Shire. Dr Li was a full-time employee of Shire at the time of the study and is now an employee of Cerexa, Inc, Oakland, California. Dr Civil is a full-time employee of Shire. Dr Paller has served as a consultant to Shire.
Funding/support: This study was funded by Shire Development, Inc, Wayne, Pennsylvania.
Acknowledgment: The authors acknowledge the study sponsor’s role in determining the study design as well as in collecting and analyzing the study data. Also, the authors acknowledge Timothy E. Wilens, MD, Substance Abuse Services, Pediatric and Adult Psychopharmacology Clinics, Massachusetts General Hospital, Boston, for his assistance with the study and manuscript. Dr Wilens has received grant/research support from and has served as a consultant to or on the speakers’ bureaus for Abbott, Eli Lilly, McNeil, Merck, National Institutes of Health (National Institute on Drug Abuse), Novartis, and Shire. Lastly, the authors acknowledge Tracey Fine, MS, ELS, and Laura Miesle, PharmD, CMPP, The JB Ashtin Group Inc, Plymouth, Michigan, for their editorial assistance in the preparation of this manuscript. This editorial assistance was funded by Shire Development, Inc.
REFERENCES
1. Bloom B, Dey AN, Freeman G. Summary health statistics for US children: National Health Interview Survey, 2005. Vital Health Stat 10. 2006;231(231):1-84. PubMed
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
3. Biederman J, Faraone S, Milberger S, et al. Predictors of persistence and remission of ADHD into adolescence: results from a four-year prospective follow-up study. J Am Acad Child Adolesc Psychiatry. 1996;35(3):343-351.PubMed doi:10.1097/00004583-199603000-00016
4. Pliszka S; AACAP Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.PubMed doi:10.1097/chi.0b013e318054e724
5. Bush G, Spencer TJ, Holmes J, et al. Functional magnetic resonance imaging of methylphenidate and placebo in attention-deficit/hyperactivity disorder during the multi-source interference task. Arch Gen Psychiatry. 2008;65(1):102-114.PubMed doi:10.1001/archgenpsychiatry.2007.16
6. Wilens TE. Effects of methylphenidate on the catecholaminergic system in attention-deficit/hyperactivity disorder. J Clin Psychopharmacol. 2008;28(suppl 2):S46-S53.PubMed doi:10.1097/JCP.0b013e318173312f
7. Biederman J, Quinn D, Weiss M, et al. Efficacy and safety of Ritalin LA, a new, once daily, extended-release dosage form of methylphenidate, in children with attention deficit hyperactivity disorder. Paediatr Drugs. 2003;5(12):833-841.PubMed doi:10.2165/00148581-200305120-00006
8. Wilens TE, Boellner SW, López FA, et al. Varying the wear time of the methylphenidate transdermal system in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2008;47(6):700-708.PubMed doi:10.1097/CHI.0b013e31816bffdf
9. McGough JJ, Wigal SB, Abikoff H, et al. A randomized, double-blind, placebo-controlled, laboratory classroom assessment of methylphenidate transdermal system in children with ADHD. J Atten Disord. 2006;9(3):476-485.PubMed doi:10.1177/1087054705284089
10. Findling RL, Bukstein OG, Melmed RD, et al. A randomized, double-blind, placebo-controlled, parallel-group study of methylphenidate transdermal system in pediatric patients with attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2008;69(1):149-159.PubMed doi:10.4088/JCP.v69n0120
11. Musel AL, Warshaw EM. Cutaneous reactions to transdermal therapeutic systems. Dermatitis. 2006;17(3):109-122. PubMed
12. Pelham WE Jr, Manos MJ, Ezzell CE, et al. A dose-ranging study of a methylphenidate transdermal system in children with ADHD. J Am Acad Child Adolesc Psychiatry. 2005;44(6):522-529.PubMed doi:10.1097/01.chi.0000157548.48960.95
13. Pelham WE, Burrows-Maclean L, Gnagy EM, et al. Transdermal methylphenidate, behavioral, and combined treatment for children with ADHD. Exp Clin Psychopharmacol. 2005;13(2):111-126.PubMed doi:10.1037/1064-1297.13.2.111
14. Findling RL, Wigal SB, Bukstein OG, et al. Long-term tolerability of the methylphenidate transdermal system in pediatric attention-deficit/hyperactivity disorder: a multicenter, prospective, 12-month, open-label, uncontrolled, phase III extension of four clinical trials. Clin Ther. 2009;31(8):1844-1855.PubMed doi:10.1016/j.clinthera.2009.08.002
15. Arnold LE, Bozzolo DR, Hodgkins P, et al. Switching from oral extended-release methylphenidate to the methylphenidate transdermal system: continued attention-deficit/hyperactivity disorder symptom control and tolerability after abrupt conversion. Curr Med Res Opin. 2010;26(1):129-137.PubMed doi:10.1185/03007990903437412
16. Daytrana [package insert]. Wayne, PA: Shire US Inc; 2008.
17. DuPaul GJ, Power TJ, Anastopoulos AD, et al. ADHD Rating Scale-IV: Checklists, Norms, and Clinical Interpretation. New York, NY: The Guilford Press; 1998.
18. Fowler JF, Warshaw EM, Squires L. Methylphenidate patch-test protocol and irritancy threshold determination in healthy adult subjects. Dermatitis. 2009;20(5):271-274. PubMed
19. Pratt MD, Belsito DV, DeLeo VA, et al. North American Contact Dermatitis Group patch-test results, 2001-2002 study period. Dermatitis. 2004;15(4):176-183. PubMed
20. National Institute of Mental Health. CGI (Clinical Global Impressions) scale. Psychopharmacol Bull. 1985;21:839-843.
21. Warshaw EM, Paller AS, Fowler JF, et al. Practical management of cutaneous reactions to the methylphenidate transdermal system: recommendations from a dermatology expert panel consensus meeting. Clin Ther. 2008;30(2):326-337.PubMed doi:10.1016/j.clinthera.2008.01.022
22. Horning JR, Zawada ET Jr, Simmons JL, et al. Efficacy and safety of two-year therapy with transdermal clonidine for essential hypertension. Chest. 1988;93(5):941-945.PubMed doi:10.1378/chest.93.5.941
23. Maibach HI. Oral substitution in patients sensitized by transdermal clonidine treatment. Contact Dermat. 1987;16(1):1-8.PubMed doi:10.1111/j.1600-0536.1987.tb02607.x
24. Confino-Cohen R, Goldberg A. Successful desensitization of methylphenidate-induced rash. J Child Adolesc Psychopharmacol. 2005;15(4):703-705.PubMed doi:10.1089/cap.2005.15.703
25. Pelham WE, Gnagy EM, Burrows-Maclean L, et al. Once-a-day Concerta methylphenidate versus three-times-daily methylphenidate in laboratory and natural settings. Pediatrics. 2001;107(6):e105-e119.PubMed doi:10.1542/peds.107.6.e105
26. Quinn D, Wigal S, Swanson J, et al. Comparative pharmacodynamics and plasma concentrations of d-threo-methylphenidate hydrochloride after single doses of d-threo-methylphenidate hydrochloride and d,l-threo-methylphenidate hydrochloride in a double-blind, placebo-controlled, crossover laboratory school study in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2004;43(11):1422-1429.PubMed doi:10.1097/01.chi.0000140455.96946.2b
27. Stein MA, Sarampote CS, Waldman ID, et al. A dose-response study of OROS methylphenidate in children with attention-deficit/hyperactivity disorder. Pediatrics. 2003;112(5):e404-e413.PubMed doi:10.1542/peds.112.5.e404
28. Swanson JM, Wigal SB, Wigal T, et al; COMACS Study Group. A comparison of once-daily extended-release methylphenidate formulations in children with attention-deficit/hyperactivity disorder in the laboratory school (the COMACS Study). Pediatrics. 2004;113(3 pt 1):e206-e216.PubMed doi:10.1542/peds.113.3.e206
29. Wolraich ML, Greenhill LL, Pelham W, et al; Concerta Study Group. Randomized, controlled trial of oros methylphenidate once a day in children with attention-deficit/hyperactivity disorder. Pediatrics. 2001;108(4):883-892.PubMed doi:10.1542/peds.108.4.883


