Objective: To determine the efficacy, safety, and tolerability of vilazodone in the treatment of posttraumatic stress disorder (PTSD) with comorbid mild-to-moderate depression.
Methods: A 12-week randomized, double-blind, placebo-controlled trial was conducted in adult outpatients who met DSM-IV criteria for PTSD with comorbid depression between February 2013 and September 2015. Participants were randomly assigned to receive vilazodone 40 mg/d or placebo, and outcome measures were obtained at scheduled visits. Primary outcome measures included change in PTSD symptoms from baseline to end of study as indexed by the Clinician-Administered PTSD Scale (CAPS) and PTSD Symptom Scale-Self-Report (PSS-SR). Secondary outcome measures of anxiety, depression, and impairment were obtained, as well as biomarker assessment at baseline and end of study.
Results: A total of 59 patients were randomly assigned to receive vilazodone (n = 29) or placebo (n = 30). Of those who were randomized, there were 25 completers in the vilazodone group and 22 completers in the placebo group. No significant differences were observed between the groups on any of the primary or secondary outcome measures. Vilazodone was generally well tolerated with few differences in the rate of adverse events between groups.
Conclusions: Treatment with vilazodone 40 mg/d did not improve symptoms of PTSD and comorbid depression. Further investigation of the biological mechanisms underlying PTSD may lead to identification of improved therapeutic targets.
Trial Registration: ClinicalTrials.gov identifier: NCT01715519
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
A Double-Blind, Placebo-Controlled Randomized Trial of Vilazodone in the Treatment of Posttraumatic Stress Disorder
and Comorbid Depression
ABSTRACT
Objective: To determine the efficacy, safety, and tolerability of vilazodone in the treatment of posttraumatic stress disorder (PTSD) with comorbid mild-to-moderate depression.
Methods: A 12-week randomized, double-blind, placebo-controlled trial was conducted in adult outpatients who met DSM-IV criteria for PTSD with comorbid depression between February 2013 and September 2015. Participants were randomly assigned to receive vilazodone 40 mg/d or placebo, and outcome measures were obtained at scheduled visits. Primary outcome measures included change in PTSD symptoms from baseline to end of study as indexed by the Clinician-Administered PTSD Scale (CAPS) and PTSD Symptom Scale–Self-Report (PSS-SR). Secondary outcome measures of anxiety, depression, and impairment were obtained, as well as biomarker assessment at baseline and end of study.
Results: A total of 59 patients were randomly assigned to receive vilazodone (n = 29) or placebo (n = 30). Of those who were randomized, there were 25 completers in the vilazodone group and 22 completers in the placebo group. No significant differences were observed between the groups on any of the primary or secondary outcome measures. Vilazodone was generally well tolerated with few differences in the rate of adverse events between groups.
Conclusions: Treatment with vilazodone 40 mg/d did not improve symptoms of PTSD and comorbid depression. Further investigation of the biological mechanisms underlying PTSD may lead to identification of improved therapeutic targets.
Trial Registration: ClinicalTrials.gov identifier: NCT01715519
Prim Care Companion CNS Disord 2017;19(4):17m02138
https://doi.org/10.4088/PCC.17m02138
© Copyright 2017 Physicians Postgraduate Press, Inc.
aDepartment of Psychiatry, Creighton University, Omaha, Nebraska
bVA Nebraska-Western Iowa Health Care System, Omaha, Nebraska
cVA Long Beach Healthcare System, Long Beach, California
dDepartment of Psychiatry and Behavioral Sciences, University of California at Irvine, Irvine, California
eCollege of Public Health, University of Nebraska Medical Center, Omaha, Nebraska
fBanner Thunderbird Medical Center, Glendale, Arizona
*Corresponding author: Sriram Ramaswamy, MD, Department of Veterans Affairs, VA Nebraska-Western Iowa Health Care System, 4101 Woolworth Ave, Omaha, NE 68105 ([email protected]).
Posttraumatic stress disorder (PTSD) is a debilitating illness characterized by re-experiencing aspects of the original trauma, avoidance and numbing of trauma reminders, and general hyperarousal. Lifetime prevalence of PTSD in community samples is around 6.8% and as high as 30% among Vietnam veterans and female victims of rape.1,2 Rates of PTSD among returning Gulf War veterans have ranged from 8% to 10%,3,4 while the prevalence of current PTSD among Operation Enduring Freedom and Operation Iraqi Freedom veterans is around 13.8%,5 with new incidence rates of 10–13 cases per 1,000 person years.6 The estimated cost for mental health–related expenses in these veterans is $4–6 billion over 2 years.5 PTSD has a high comorbidity (83%–90%) with other psychiatric disorders, including mood, substance use, personality, and panic disorders.7 Approximately 50%–60% of PTSD patients have major depressive disorder.7 These disorders are associated with significant social and interpersonal impairment.
The evidence base for pharmacologic treatment is strongest for the selective serotonin reuptake inhibitors (SSRIs). The only 2 US Food and Drug Administration–approved medications for the treatment of PTSD are sertraline and paroxetine. Vilazodone is a combination SSRI and a partial agonist of serotonergic (5-HT1A) receptors. Vilazodone has demonstrated clinical antidepressant efficacy equivalent to SSRIs,8–12 and there is recent evidence13,14 that it may be an effective treatment for anxiety disorders. Sleep disturbances are a hallmark symptom in PTSD, and common polysomnographic findings include increased light sleep, reduced slow-wave sleep, and increased rapid eye movement density.15 In early phase I studies,16 vilazodone completely abolished rapid eye movement sleep. Vilazodone’s unique profile suggests potential anxiolytic and antidepressant effects and specific benefit for sleep and traumatic nightmares, which are critical to assist those suffering from PTSD. The primary aim of this study was to assess the safety and efficacy of vilazodone for treatment of PTSD.
There is great interest in identifying biomarkers that might serve as predictors of treatment response in PTSD. PTSD is characterized by hypothalamic-pituitary-adrenal axis dysfunction and possibly chronic inflammation, which can be identified using assays such as C-reactive protein (CRP),17 salivary cortisol,18–21 and 24-hour urine catecholamines.18,19,22 These biomarkers have been studied in individuals with PTSD23,24 and were included in this study to further examine their relationship to treatment response in veterans with PTSD.
METHODS
Study Design
The study was a 12-week prospective, double-blind, placebo-controlled trial of vilazodone 40 mg for PTSD and comorbid depression. The trial was conducted between February 2013 and September 2015 at 2 sites: the VA Long Beach Healthcare System (Long Beach, California) and the VA Nebraska-Western Iowa Healthcare System (Omaha, Nebraska). The study was approved by the institutional review boards at both sites and was registered at ClinicalTrials.gov (identifier: NCT01715519). All participants provided written informed consent.
Participants
Recruitment was conducted at both sites by posting of flyers, outreach to mental health clinics, and word of mouth. The study included outpatient men and women aged 18 to 55 years who were diagnosed with chronic PTSD using DSM-IV criteria for chronic PTSD with a Clinician-Administered PTSD Scale (CAPS)25 score ≥ 50 and who had at least mild depression on the Beck Depression Inventory-II (BDI-II score ≥ 12).26 Eligible persons were allowed to have other symptoms that are commonly comorbid with PTSD and could be engaged in supportive psychotherapy if it was initiated at least 3 months prior to the screening visit or if it followed a standard 12-week evidence-based psychotherapy such as prolonged exposure or cognitive processing therapy and if the potential subject agreed not to alter therapy during the study.
Exclusion criteria included any cognitive, psychotic, severe mood, or substance use disorder; decisional incapacity; centrally acting medications that potentially have an effect on biological expression; chronic pain levels requiring use of any opiate medications; known exposure to chemicals or physical traumas that cause neuropsychiatric sequelae (eg, traumatic brain injury); past chronic PTSD from events that preceded the incident traumatic event responsible for the current PTSD; 2 or more treatment failures on SSRIs given primarily for the treatment of PTSD; a history of seizures; a significant risk of suicide or homicide; treatment with electroconvulsive therapy within 3 months prior to screening; women who are pregnant or nursing or women of childbearing potential who are sexually active and who do not use adequate contraception; a positive urine drug screen; clinically significant medical conditions; concomitant treatment with any psychotropic drug (except zolpidem for sleep); initiation or termination of any form of psychotherapy during the study; and receiving disability payments or those who are involved in litigation for PTSD or other psychiatric illnesses.
Study Procedures
The study schedule included a screening/diagnostic visit, baseline visit, 4 treatment visits (weeks 1, 2, 4, and 8), and an end-of-study/early termination visit (week 12), followed by a 2-week taper phase (weeks 13 and 14) and safety follow-up visit (week 16). Screening was a 2-phase process. The research coordinator at each site administered the Mini-International Neuropsychiatric Interview27 to assess for comorbid and exclusionary conditions. Co-principal investigators (PIs) conducted the CAPS and medical assessments. Randomization was conducted by the study pharmacist using a computer-generated random number generator at each site. All other study personnel and subjects remained blind to group allocation until study completion.
Intervention Protocol
Participants were randomly assigned to daily doses of vilazodone 40 mg or matching placebo. The titration schedule was as follows: 10 mg/d on days 1 to 7, 20 mg/d on days 8 to 14, and 40 mg/d week 3 to end week 12. Subjects were tapered off vilazodone as follows: 20 mg/d week 13, 10 mg/d week 14, and no medication during week 15. Both study medication and placebo were administered as a single daily dose in the morning. Later during the trial, at the discretion of the PIs, dosing may have been switched to either the morning or evening dependent on individual subject side effects. Subjects were instructed to take study medication or placebo with food. During the course of the trial, the PIs had the option to adjust the dosage regimen based on the patient’s response, including the Clinical Global Impressions scale (CGI)28 and the self-rating scales assessment as well as tolerance to study medication. The maximum dose permitted for vilazodone was 40 mg/d. Treatment compliance was monitored by counts of returned medication. Any subject with ongoing adverse events or abnormal findings on final physical examination or laboratory assessment was followed until the condition returned to pretrial status or could be explained as being unrelated to the study drug.
Outcome Measures
Primary outcomes were change in mean total scores on the CAPS and the PTSD Symptom Scale–Self-Report (PSS-SR)29 and change in mean sleep scores on the CAPS (items 2 and 13), PSS-SR (item 13), and BDI-II (item 16) from baseline to end of study/early termination.
Secondary outcomes were change in mean scores for depression, anxiety, and impairment as rated by the BDI-II, the Hamilton Anxiety Scale (HARS),30 the CGI, and the Sheehan Disability Scale (SDS)31 from baseline to end of study/early termination.
Biomarkers were collected at baseline and end of study/early termination visits. CRP was assessed from serum taken in the morning. Cortisol was collected between 8 am and 9 am or within 2 hours of waking with self-administered salivary tubes. The 24-hour urine catecholamines (epinephrine, norepinephrine, and dopamine) were collected by wasting the first morning urine and then collecting all urine for 24 hours including the second morning urine. Samples were brought to the laboratory the day of final collection and were kept refrigerated until delivery.
Safety and tolerability were assessed by adverse events and vital signs recorded at each visit and laboratory tests conducted at baseline and at end of study. Suicidality was assessed at baseline and week 4, 8, and 12 visits using the Columbia-Suicide Severity Rating Scale (C-SSRS).32 The Changes in Sexual Functioning Questionnaire (CSFQ)33 was also administered at baseline and weeks 4, 8, and 12.
Data Management and Analyses
Baseline characteristics were compared between sites using t tests for continuous variables and χ2 or Fisher exact test for categorical variables. Analysis of variance was used to look at the change in measures by randomization group, adjusting for site. The Tukey method was used to adjust for multiple comparisons. Linear regression was used to look at the association between change in CAPS total scores and change in biomarkers, adjusting for site. C-SSRS safety results were compared between groups using the Cochran-Mantel-Haenszel test, adjusting for site. P values less than .05 are considered statistically significant. SAS software version 9.3 was used for data analysis (SAS Institute Inc, Cary, North Carolina).
RESULTS
Sample Characteristics
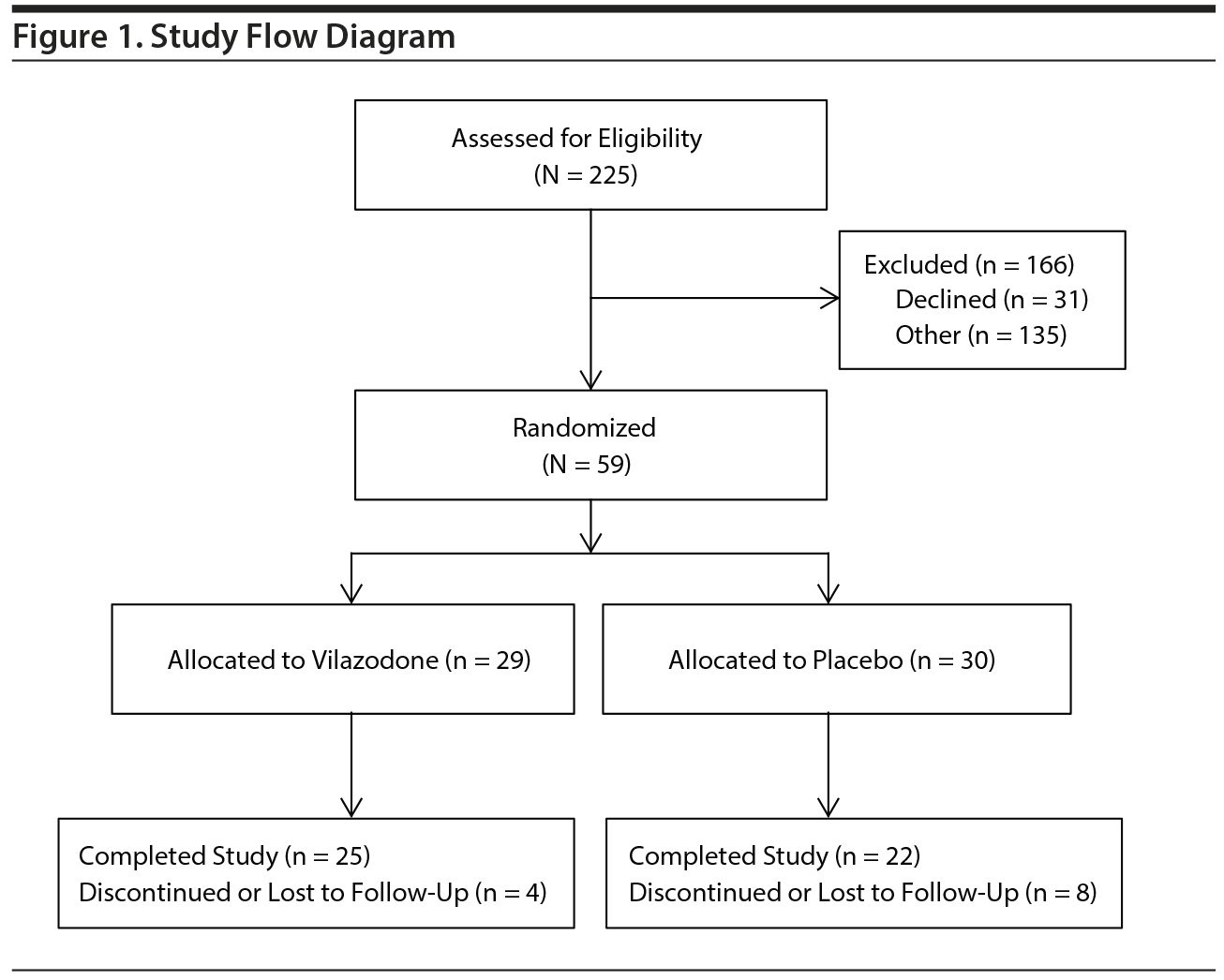
Recruitment flow across both study sites is shown in Figure 1. A total of 225 veterans were contacted due to study interest and were screened. Thirty-one elected not to proceed, and 135 were screen failures for various reasons. Fifty-nine were randomized: 29 to vilazodone and 30 to placebo. Twelve participants discontinued or were lost to follow-up, leaving 47 treatment completers.
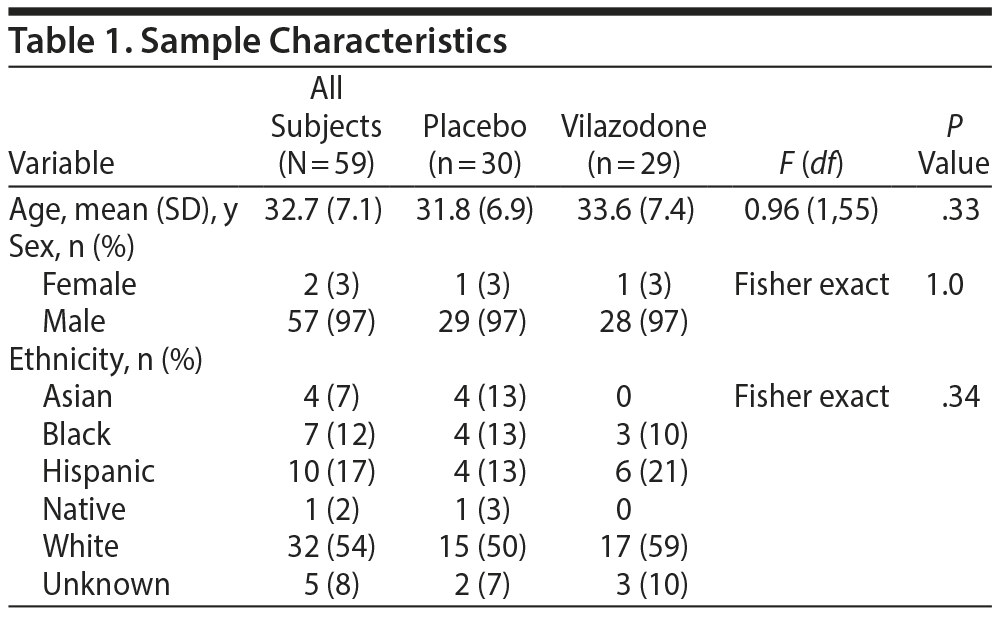
Baseline characteristics of the sample are reported in Table 1. The groups did not differ significantly by age, sex, or ethnicity (P > .3).
Primary Outcomes
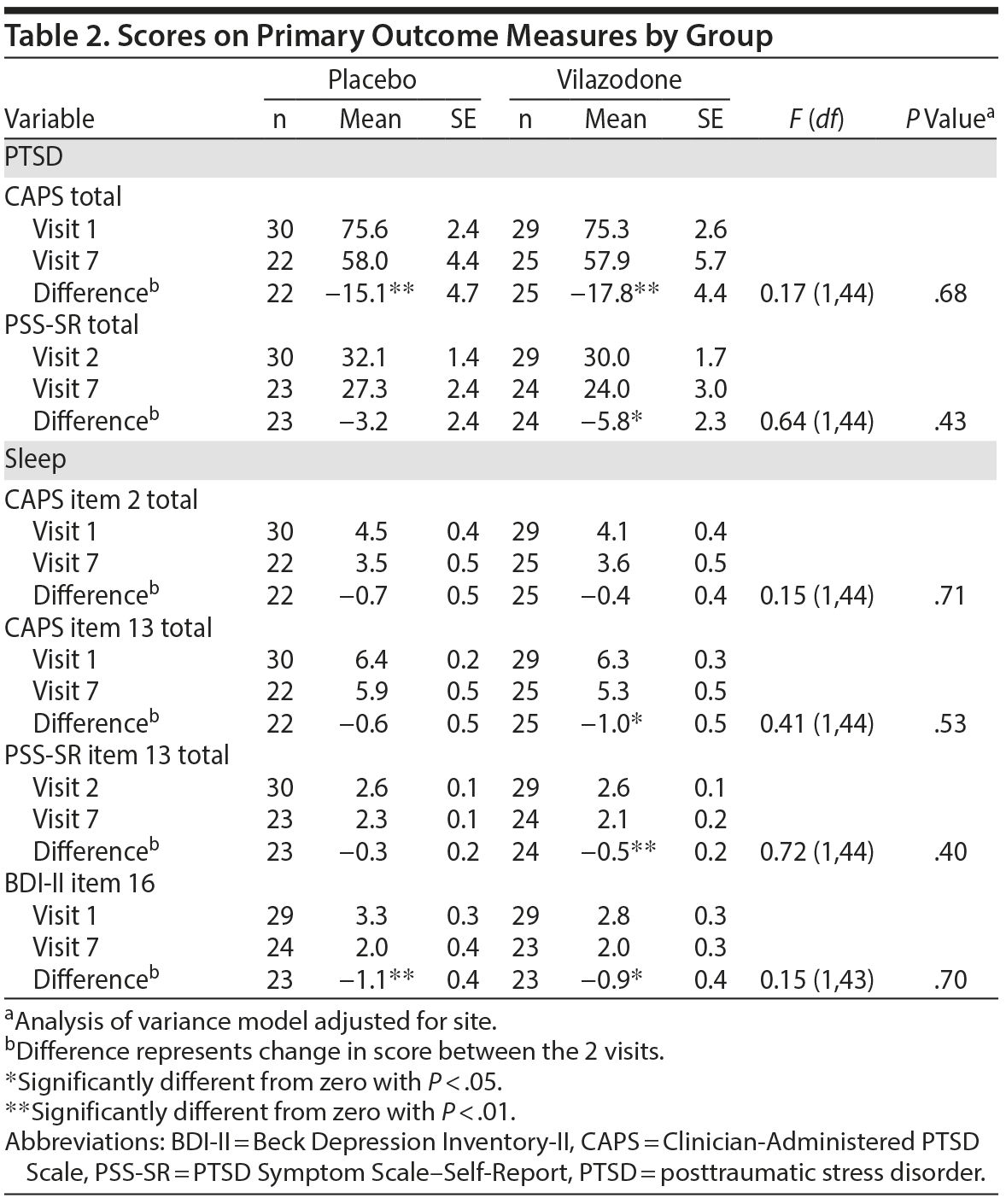
Table 2 summarizes scores on primary outcome measures of PTSD (CAPS and PSS-SR) and sleep (CAPS items 2 and 13, PSS-SR item 13, BDI-II item 16) by group from baseline to end of study. Both the vilazodone and placebo groups showed significant reductions in CAPS total score (P < .01), but no significant difference was found between groups after adjusting for site (P = .68). On the PSS-SR, the vilazodone group showed a significant decrease from visit 2 to visit 7 (P < .05). However, there was no significant difference observed between the 2 groups (P = .43). No significant group differences were observed on the sleep measures (all P > 0.1), although each group showed significant reductions on individual sleep measures from baseline to end of study (see Table 2).
Secondary Outcomes
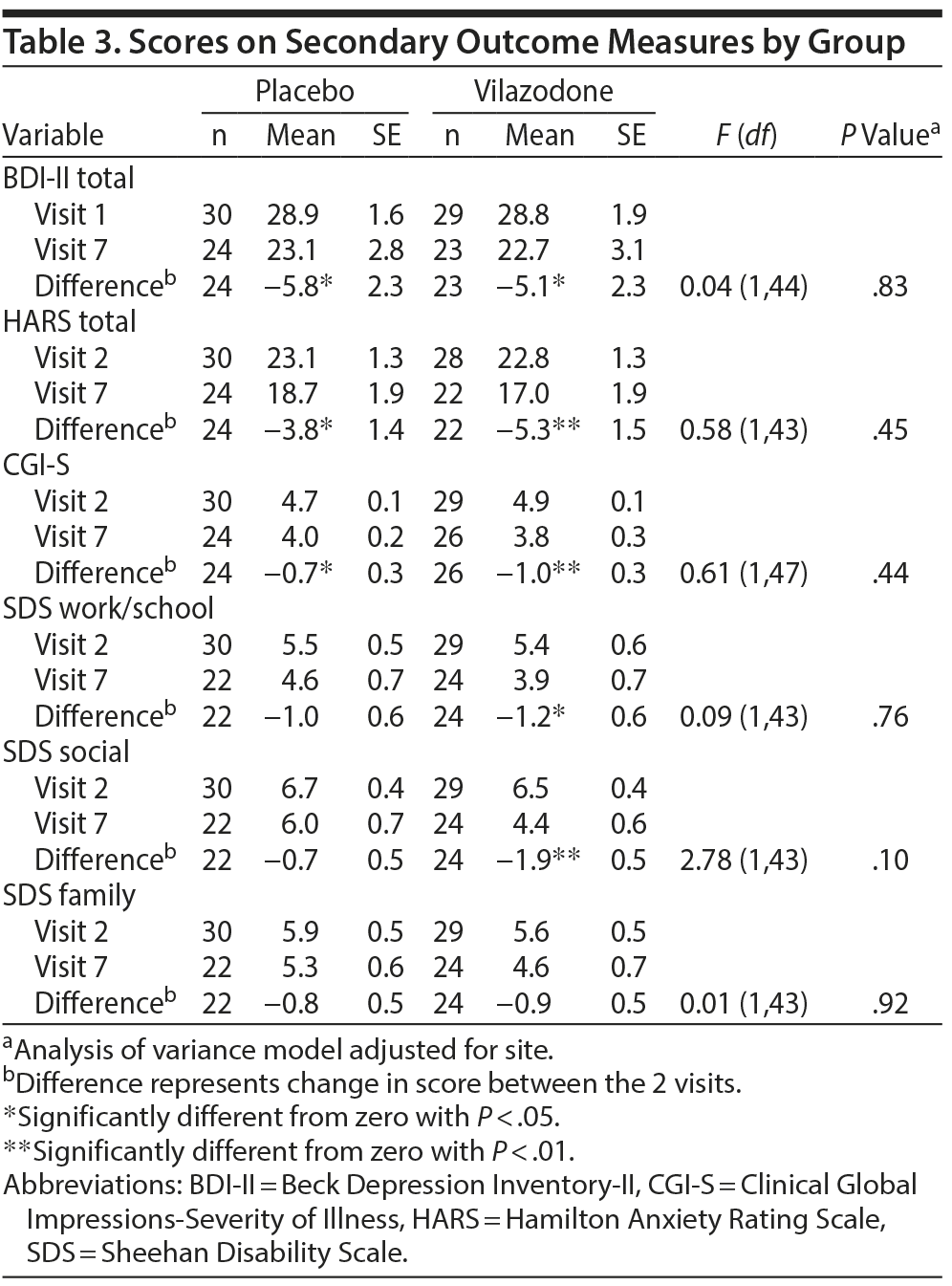
Changes on measures of depression, anxiety, and impairment are summarized in Table 3. Both groups showed significant reductions from visit 1 to 7 on the BDI-II, HARS, and CGI-S (P < .05). In addition, the vilazodone group showed a significant decrease in SDS work/school and social life scores (P < .05). However, no significant group differences were observed on any of these measures.
Biomarker Outcomes
Table 4 summarizes changes in biomarkers from baseline to end of study/early termination, including CRP, salivary cortisol, and urinary catecholamines. No significant effects were detected for CRP or cortisol. In addition, the vilazodone group but not the placebo group showed significant reductions in norepinephrine, dopamine, epinephrine, and total catecholamines between visit 1 and visit 7 (P < .05), although there were no significant group differences on these measures.
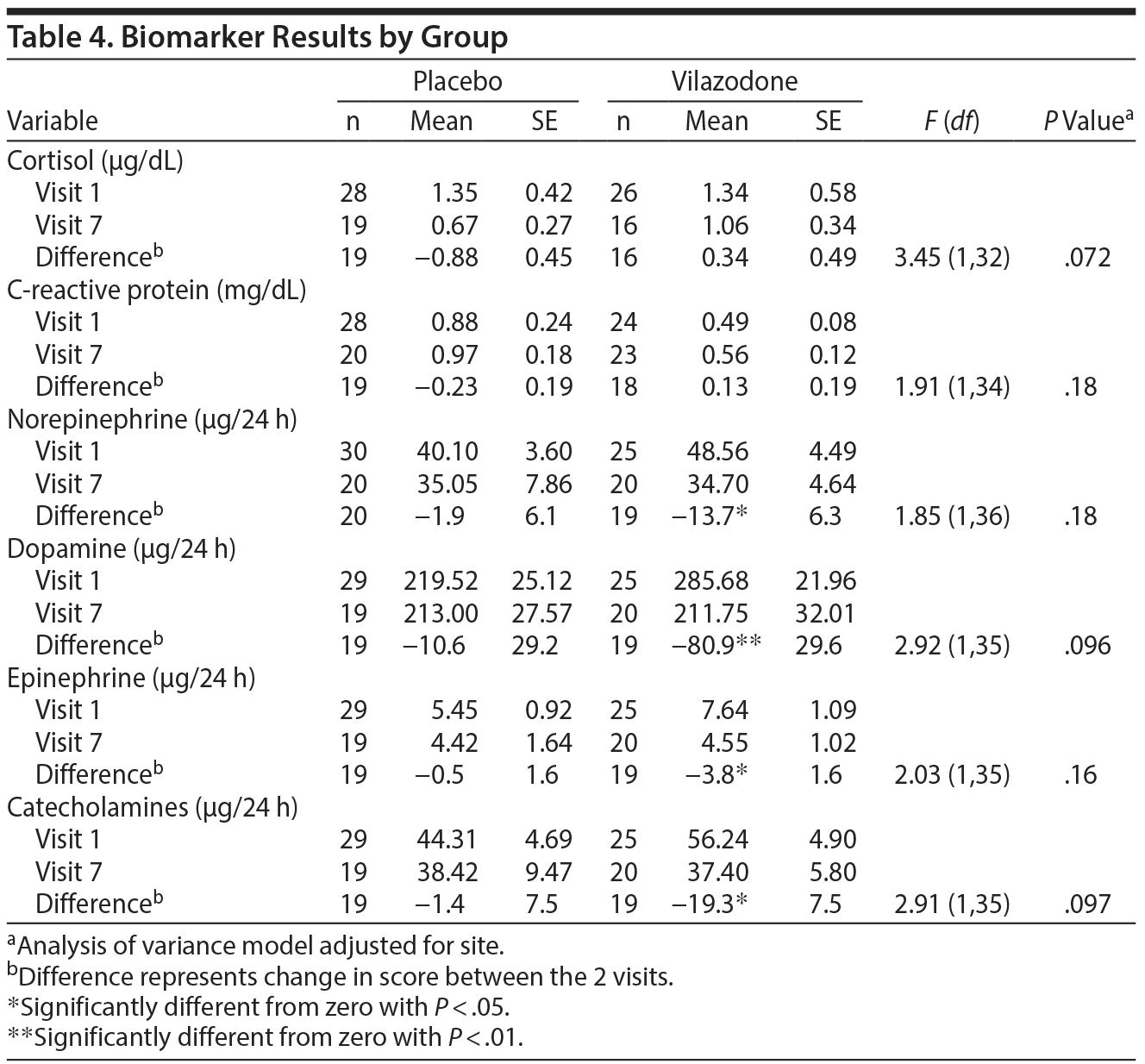
Linear regression analyses were also conducted to examine associations between change in biomarker measures and CAPS total scores from baseline to end of study. Change in epinephrine was found to significantly predict change in CAPS total scores (β = 1.01, P = .031), and change in total catecholamines was a marginally significant predictor of change in CAPS total scores (β = .19, P = .065).
Safety and Tolerability
The most commonly reported adverse events in the sample were gastrointestinal, sexual, and sleep-related events; headaches; and irritability. The rate of gastrointestinal events was higher in the vilazodone group at visit 3 (P < .05), although no other differences in side effects were observed. On the C-SSRS, the proportion of patients endorsing suicidal ideation or behavior during the study was relatively low (range, 0%–20.8%), and no differences were observed between the groups. However, there were 2 serious adverse events reported at 1 site (suicidal ideation and self-mutilation) in 1 patient in the vilazodone study group. This patient was discontinued from the trial.
In addition, participants in the vilazodone group showed a significant decrease in CSFQ total scores from baseline to end of study (P > .05), although no difference was observed between the groups (P = .36).
DISCUSSION
This is the first study evaluating the efficacy, safety, and tolerability of vilazodone in the treatment of PTSD with comorbid mild-to-moderate depression. Vilazodone was generally well tolerated in our sample with few reported side effects. However, no significant differences were observed between groups on the primary or secondary outcome measures. These results are similar to those of previous clinical trials34–37 of SSRIs in veterans with combat-related PTSD.
There are a number of factors that may have contributed to the negative findings. First, the ability to detect differences between groups was limited by the small sample size. Second, our study sample was a mixed state of depression, anxiety, and PTSD depression, which could have contributed to the lack of efficacy. It is possible that a longer duration of treatment and excluding patients with major depressive disorder might have resulted in a different outcome.38–40 Also, as medication dose and taper were set by the protocol unlike routine clinical practice, it is possible that our subjects might have responded to smaller doses and a slower titration. Third, it has been suggested that patients seeking or receiving compensation for PTSD may be reluctant to report improvement in symptoms, although evidence of such an effect has been mixed.41,42 As we excluded individuals seeking or receiving greater than 50% service-connected or other disability for PTSD, it is unlikely that such an effect played a significant role in this study. Finally, placebo response in psychiatric clinical trials has been well documented,43–45 although there are data40 to suggest placebo worsens symptoms in patients with combat-related PTSD. The use of a double-blind design and standardized measures reduced the potential for bias in this study, although there are other factors (eg, patient expectancy of improvement, benefit of contact with health care providers) that may have reduced our ability to detect differences between drug and placebo. Methods that may help to minimize the impact of placebo response, such as use of a placebo lead-in, should be considered in the design of future studies.
It has been suggested that combat-related PTSD might be resistant to SSRIs.46,47 The lack of efficacy of vilazodone might be similar to other negative studies48 with SSRIs in PTSD: nonspecificity of SSRIs and failure to extrapolate the serotonergic deficiency hypothesis of mood and anxiety disorders to PTSD. Despite vilazodone’s potent anxiolytic effect in patients with anxious depression,49 our study failed to demonstrate an effect on anxiety symptoms. Since PTSD typically presents with comorbid anxiety and mood disorders, exclusion of patients with PTSD in the pivotal vilazodone studies might explain this anomaly. There is evidence from animal models of chronic stress50 and clinical studies of GAD and depression51 for an alerted 5-HT1A receptor expression, but vilazodone 5-HT1A agonistic actions did not prove to be beneficial in our study sample of combat veterans with PTSD.
Given the overall lackluster effect of SSRIs in veterans, the theory of an exaggerated noradrenergic system in PTSD52 has assumed greater importance, especially against the backdrop of robust data53 with prazosin in combat veterans with PTSD. In our study, the vilazodone group showed significant reductions in norepinephrine, epinephrine, and total catecholamines between visit 1 and visit 7, change in epinephrine was found to significantly predict change in CAPS total scores, and change in total catecholamines was a marginally significant predictor of change in CAPS total scores. The neurobiology of PTSD is complex and involves multiple neurotransmitter systems. The serotonin system is believed to interact with the norepinephrine and corticotrophin-releasing hormone systems in coordinating affective and stress responses.54,55 It is plausible that a longer duration of treatment with vilazodone could have induced downstream effects and a possible therapeutic response. To our knowledge, this is the first study that attempted to correlate SSRI response in combat PTSD to catecholamine markers. Future studies looking at treatment approaches that simultaneously and synergistically target both serotonergic and noradrenergic systems might provide useful information.
There is a general notion that it is difficult to demonstrate a treatment effect with pharmaceutical therapies in clinical trials involving combat veterans with PTSD. However, this does not appear to be entirely true when one looks at studies with the α1-adrenergic receptor blocker prazosin in PTSD. Multiple randomized controlled trials have consistently demonstrated the efficacy of prazosin for nightmares and sleep disturbances and improving overall PTSD symptoms and functionality.56 Also, a recent monotherapy study57 with the sedating antipsychotic quetiapine found it effective in the treatment of PTSD and associated depression and anxiety symptoms. Our study found no differences on sleep measures between the 2 groups. It is plausible that early restoration of sleep disturbances in PTSD is critical for recovery from PTSD symptoms, and future studies with SSRIs should examine this important mediator of treatment in more detail.
In conclusion, this study did not find vilazodone monotherapy to be effective in the treatment of PTSD in a group of young, predominantly male combat veterans, consistent with previous work34–37 suggesting limited efficacy of SSRIs for treatment of PTSD. The failure of such approaches might be attributable to lack of clear delineation of the different neurotransmitter and neuroanatomical pathways involved in the pathophysiology of PTSD.58 While designing future studies in PTSD, careful attention to strategies that maximize and enhance signal detection should be considered.59 Current Department of Veterans Affairs/Department of Defense clinical practice guidelines60 recommend SSRIs (fluoxetine, paroxetine, or sertraline) or serotonin-norepinephrine reuptake inhibitors (venlafaxine) as first-line treatment for PTSD. Second-line agents include mirtazapine, nefazodone, tricyclic antidepressants (amitriptyline and imipramine), or monoamine oxidase inhibitors (phenelzine). Following negative results from the Veterans Affairs–sponsored study61 of risperidone in PTSD, the guidelines60 were revised to recommend against the use of adjunctive risperidone in PTSD. Given the growing body of literature suggesting the limited efficacy of SSRIs in combat PTSD, one might be tempted to reexamine their utility in PTSD; however, given that benefits of SSRIs substantially outweigh harm, it is very likely that clinicians will continue to prescribe SSRIs for PTSD.
Submitted: March 16, 2017; accepted May 26, 2017.
Published online: August 24, 2017.
Potential conflicts of interest: None.
Funding/support: This work was supported by Forest Laboratories, Inc.
Role of the sponsor: Forest Laboratories, Inc had no role in the design, analysis, interpretation, or publication of this study.
REFERENCES
1. Kulka RA, Schlenger WE, Fairbank JA, et al. National Vietnam Veterans Readjustment Study (NVVRS): Description, Current Status, and Initial PTSD Prevalence Estimates. Research Triangle Park, NC: Research Triangle Institute; 1988.
2. Resnick HS, Kilpatrick DG, Dansky BS, et al. Prevalence of civilian trauma and posttraumatic stress disorder in a representative national sample of women. J Consult Clin Psychol. 1993;61(6):984–991. PubMed doi:10.1037/0022-006X.61.6.984
3. Kang HK, Natelson BH, Mahan CM, et al. Posttraumatic stress disorder and chronic fatigue syndrome–like illness among Gulf War veterans: a population-based survey of 30,000 veterans. Am J Epidemiol. 2003;157(2):141–148. PubMed doi:10.1093/aje/kwf187
4. Stretch RH, Marlowe DH, Wright KM, et al. Posttraumatic stress disorder symptoms among Gulf War veterans. Mil Med. 1996;161(7):407–410. PubMed
5. Tanielian T, Jaycox LH. Invisible Wounds of War: Psychological and Cognitive Injuries, Their Consequences, and Services to Assist Recovery. Santa Monica, CA: RAND Corporation; 2008.
6. Smith TC, Ryan MA, Wingard DL, et al; Millennium Cohort Study Team. New onset and persistent symptoms of posttraumatic stress disorder self-reported after deployment and combat exposures: prospective population-based US military cohort study. BMJ. 2008;336(7640):366–371. PubMed doi:10.1136/bmj.39430.638241.AE
7. Bradley R, Greene J, Russ E, et al. A multidimensional meta-analysis of psychotherapy for PTSD. Am J Psychiatry. 2005;162(2):214–227. PubMed doi:10.1176/appi.ajp.162.2.214
8. Rickels K, Athanasiou M, Robinson DS, et al. Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(3):326–333. PubMed doi:10.4088/JCP.08m04637
9. Viibryd (vilazodone) [package insert]. Jersey City, NJ: Forest Laboratories; 2011.
10. Laughren TP, Gobburu J, Temple RJ, et al. Vilazodone: clinical basis for the US Food and Drug Administration’s approval of a new antidepressant. J Clin Psychiatry. 2011;72(9):1166–1173. PubMed doi:10.4088/JCP.11r06984
11. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643–646. PubMed
12. Croft HA, Pomara N, Gommoll C, et al. Efficacy and safety of vilazodone in major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2014;75(11):e1291–e1298. PubMed doi:10.4088/JCP.14m08992
13. Gommoll C, Forero G, Mathews M, et al. Vilazodone in patients with generalized anxiety disorder: a double-blind, randomized, placebo-controlled, flexible-dose study. Int Clin Psychopharmacol. 2015;30(6):297–306. PubMed doi:10.1097/YIC.0000000000000096
14. Careri JM, Draine AE, Hanover R, et al. A 12-week double-blind, placebo-controlled, flexible-dose trial of vilazodone in generalized social anxiety disorder. Prim Care Companion CNS Disord. 2015;17(6):doi:10.4088/PCC.15m01831. PubMed doi:10.4088/PCC.15m01831
15. Kobayashi I, Boarts JM, Delahanty DL. Polysomnographically measured sleep abnormalities in PTSD: a meta-analytic review. Psychophysiology. 2007;44(4):660–669. PubMed doi:10.1111/j.1469-8986.2007.537.x
16. Murck H, Frieboes RM, Antonijevic IA, et al. Distinct temporal pattern of the effects of the combined serotonin-reuptake inhibitor and 5-HT1A agonist EMD 68843 on the sleep EEG in healthy men. Psychopharmacology (Berl). 2001;155(2):187–192. PubMed doi:10.1007/s002130100703
17. von Känel R, Hepp U, Kraemer B, et al. Evidence for low-grade systemic proinflammatory activity in patients with posttraumatic stress disorder. J Psychiatr Res. 2007;41(9):744–752. PubMed doi:10.1016/j.jpsychires.2006.06.009
18. Glover DA, Poland RE. Urinary cortisol and catecholamines in mothers of child cancer survivors with and without PTSD. Psychoneuroendocrinology. 2002;27(7):805–819. PubMed doi:10.1016/S0306-4530(01)00081-6
19. Rohleder N, Karl A. Role of endocrine and inflammatory alterations in comorbid somatic diseases of posttraumatic stress disorder. Minerva Endocrinol. 2006;31(4):273–288. PubMed
20. Thaller V, Vrkljan M, Hotujac L, et al. The potential role of hypocortisolism in the pathophysiology of PTSD and psoriasis. Coll Antropol. 1999;23(2):611–619. PubMed
21. Yehuda R, Southwick SM, Krystal JH, et al. Enhanced suppression of cortisol following dexamethasone administration in posttraumatic stress disorder. Am J Psychiatry. 1993;150(1):83–86. PubMed doi:10.1176/ajp.150.1.83
22. Yehuda R, Siever LJ, Teicher MH, et al. Plasma norepinephrine and 3-methoxy-4-hydroxyphenylglycol concentrations and severity of depression in combat posttraumatic stress disorder and major depressive disorder. Biol Psychiatry. 1998;44(1):56–63. PubMed doi:10.1016/S0006-3223(98)80007-3
23. Eraly SA, Nievergelt CM, Maihofer AX, et al; Marine Resiliency Study Team. Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry. 2014;71(4):423–431. PubMed doi:10.1001/jamapsychiatry.2013.4374
24. Spitzer C, Barnow S, Völzke H, et al. Association of posttraumatic stress disorder with low-grade elevation of C-reactive protein: evidence from the general population. J Psychiatr Res. 2010;44(1):15–21. PubMed doi:10.1016/j.jpsychires.2009.06.002
25. Blake DD, Weathers FW, Nagy LM, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress. 1995;8(1):75–90. PubMed doi:10.1002/jts.2490080106
26. Beck AT, Steer RA, Ball R, et al. Comparison of Beck Depression Inventories -IA and -II in psychiatric outpatients. J Pers Assess. 1996;67(3):588–597. PubMed doi:10.1207/s15327752jpa6703_13
27. Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(suppl 20):22–33, quiz 34–57. PubMed
28. Guy W. Clinical Global Impressions (028-CGI). ECDEU Assessment Manual for Psychopharmacology Revised (DHEW Publication ADM 76-338). Rockville, MD: National Institute of Mental Health; 1976:217–222.
29. Foa EB, Riggs DS, Dancu CV, et al. Reliability and validity of a brief instrument for assessing posttraumatic stress disorder. J Trauma Stress. 1993;6:459–473. doi:10.1002/jts.2490060405
30. Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959;32(1):50–55. PubMed doi:10.1111/j.2044-8341.1959.tb00467.x
31. Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996;11(suppl 3):89–95. PubMed doi:10.1097/00004850-199606003-00015
32. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. PubMed doi:10.1176/appi.ajp.2011.10111704
33. Clayton AH, McGarvey EL, Clavet GJ. The Changes in Sexual Functioning Questionnaire (CSFQ): development, reliability, and validity. Psychopharmacol Bull. 1997;33(4):731–745. PubMed
34. Friedman MJ, Marmar CR, Baker DG, et al. Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. J Clin Psychiatry. 2007;68(5):711–720. PubMed doi:10.4088/JCP.v68n0508
35. Hertzberg MA, Feldman ME, Beckham JC, et al. Lack of efficacy for fluoxetine in PTSD: a placebo-controlled trial in combat veterans. Ann Clin Psychiatry. 2000;12(2):101–105. PubMed doi:10.3109/10401230009147096
36. van der Kolk BA, Dreyfuss D, Michaels M, et al. Fluoxetine in posttraumatic stress disorder. J Clin Psychiatry. 1994;55(12):517–522. PubMed
37. Zohar J, Amital D, Miodownik C, et al. Double-blind placebo-controlled pilot study of sertraline in military veterans with posttraumatic stress disorder. J Clin Psychopharmacol. 2002;22(2):190–195. PubMed doi:10.1097/00004714-200204000-00013
38. Davidson J, Pearlstein T, Londborg P, et al. Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: results of a 28-week double-blind, placebo-controlled study. Am J Psychiatry. 2001;158(12):1974–1981. PubMed doi:10.1176/appi.ajp.158.12.1974
39. Londborg PD, Hegel MT, Goldstein S, et al. Sertraline treatment of posttraumatic stress disorder: results of 24 weeks of open-label continuation treatment. J Clin Psychiatry. 2001;62(5):325–331. PubMed doi:10.4088/JCP.v62n0503
40. Martenyi F, Brown EB, Zhang H, et al. Fluoxetine v. placebo in prevention of relapse in posttraumatic stress disorder. Br J Psychiatry. 2002;181:315–320. PubMed doi:10.1192/bjp.181.4.315
41. Fontana A, Rosenheck R. Psychological benefits and liabilities of traumatic exposure in the war zone. J Trauma Stress. 1998;11(3):485–503. PubMed doi:10.1023/A:1024452612412
42. Committee on Veterans’ Compensation for Posttraumatic Stress Disorder; Institute of Medicine. (US) Board on Military and Veterans Health; National Research Council (US) Board on Behavioral, Cognitive, and Sensory Sciences. PTSD Compensation and Military Service. Washington, DC: National Academies Press; 2007.
43. Fava M, Evins AE, Dorer DJ, et al. The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach. Psychother Psychosom. 2003;72(3):115–127. PubMed doi:10.1159/000069738
44. Walsh BT, Seidman SN, Sysko R, et al. Placebo response in studies of major depression: variable, substantial, and growing. JAMA. 2002;287(14):1840–1847. PubMed doi:10.1001/jama.287.14.1840
45. Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. 2013;170(7):723–733. PubMed doi:10.1176/appi.ajp.2012.12040474
46. Stein MB, Kline NA, Matloff JL. Adjunctive olanzapine for SSRI-resistant combat-related PTSD: a double-blind, placebo-controlled study. Am J Psychiatry. 2002;159(10):1777–1779. PubMed doi:10.1176/appi.ajp.159.10.1777
47. Celik C, Ozdemir B, Ozmenler KN, et al. Efficacy of paroxetine and amitriptyline in combat-related posttraumatic stress disorder: an open-label comparative study. Bull Clin Psychopharmacol. 2011;21(3):179–185. doi:10.5455/bcp.20110627111141
48. Friedman MJ. Toward rational pharmacotherapy for posttraumatic stress disorder: reprise. Am J Psychiatry. 2013;170(9):944–946. PubMed doi:10.1176/appi.ajp.2013.13060768
49. Thase ME, Chen D, Edwards J, et al. Efficacy of vilazodone on anxiety symptoms in patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(6):351–356. PubMed doi:10.1097/YIC.0000000000000045
50. Toth M. 5-HT1A receptor knockout mouse as a genetic model of anxiety. Eur J Pharmacol. 2003;463(1–3):177–184. PubMed doi:10.1016/S0014-2999(03)01280-9
51. Millan MJ. The neurobiology and control of anxious states. Prog Neurobiol. 2003;70(2):83–244. PubMed doi:10.1016/S0301-0082(03)00087-X
52. Southwick SM, Bremner JD, Rasmusson A, et al. Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol Psychiatry. 1999;46(9):1192–1204. PubMed doi:10.1016/S0006-3223(99)00219-X
53. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003–1010. PubMed doi:10.1176/appi.ajp.2013.12081133
54. Ressler KJ, Nemeroff CB. Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety. 2000;12(suppl 1):2–19. PubMed doi:10.1002/1520-6394(2000)12:1+<2::AID-DA2>3.0.CO;2-4
55. Sherin JE, Nemeroff CB. Posttraumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin Neurosci. 2011;13(3):263–278. PubMed
56. Singh B, Hughes AJ, Mehta G, et al. Efficacy of prazosin in posttraumatic stress disorder: a systematic review and meta-analysis. Prim Care Companion CNS Disord. 2016;18(4):doi:10.4088/PCC.16r01943. PubMed doi:10.4088/PCC.16r01943
57. Villarreal G, Hamner MB, Cañive JM, et al. Efficacy of quetiapine monotherapy in posttraumatic stress disorder: a randomized, placebo-controlled trial. Am J Psychiatry. 2016;173(12):1205–1212. PubMed doi:10.1176/appi.ajp.2016.15070967
58. Puetz TW, Youngstedt SD, Herring MP. Effects of pharmacotherapy on combat-related PTSD, anxiety, and depression: a systematic review and meta-regression analysis. PLoS One. 2015;10(5):e0126529. PubMed doi:10.1371/journal.pone.0126529
59. Leon AC, Davis LL. Enhancing clinical trial design of interventions for posttraumatic stress disorder. J Trauma Stress. 2009;22(6):603–611. PubMed
60. The Management of Post-Traumatic Stress Working Group. VA/DoD Clinical Practice Guideline for the Management of Post-Traumatic Stress: Guideline Summary. Washington, DC: Department of Veterans Affairs and Department of Defense; 2010.
61. Krystal JH, Rosenheck RA, Cramer JA, et al; Veterans Affairs Cooperative Study No. 504 Group. Adjunctive risperidone treatment for antidepressant-resistant symptoms of chronic military service-related PTSD: a randomized trial. JAMA. 2011;306(5):493–502. PubMed doi:10.1001/jama.2011.108
Please sign in or purchase this PDF for $40.00.

