Objective: To compare esketamine to placebo, each in addition to standard-of-care treatment, for rapidly reducing major depressive disorder symptoms, including suicidal ideation.
Methods: This phase 3, double-blind, multicenter study (ASPIRE I), conducted between June 2017 and December 2018, enrolled 226 adults having major depressive disorder based on Diagnostic and Statistical Manual of Mental Disorders fifth edition (DSM-5) criteria, active suicidal ideation with intent, and need for psychiatric hospitalization. Patients were randomized 1:1 to esketamine 84 mg or placebo nasal spray twice-weekly for 4 weeks, each with comprehensive standard-of-care treatment (initial psychiatric hospitalization and newly initiated or optimized oral antidepressant[s] therapy). Change from baseline to 24 hours post-first dose in Montgomery-Asberg Depression Rating Scale (MADRS) total score (primary endpoint) was analyzed using analysis of covariance (ANCOVA), and change in Clinical Global Impression of Severity of Suicidality Revised version (CGI-SS-r; key secondary endpoint) score was analyzed using ANCOVA on ranks with treatment difference estimated using the Hodges-Lehmann estimate.
Results: Greater improvement in MADRS total score was observed with esketamine + standard-of-care versus placebo + standard-of-care at 24 hours (least-squares mean difference [SE]: −3.8 [1.39]; 95% CI, −6.56 to −1.09; 2-sided P = .006), as well as at earlier (4 hours) and later time points during 4-week double-blind treatment. The difference between groups in the severity of suicidality was not statistically significant (median of treatment difference [95% CI]: 0.0 [−1.00 to 0.00]; 2-sided P = .107). The most common adverse events among esketamine-treated patients were dizziness, dissociation, headache, nausea, and somnolence.
Conclusions: These findings demonstrate rapid and robust efficacy of esketamine nasal spray in reducing depressive symptoms in severely ill patients with major depressive disorder who have active suicidal ideation with intent.
Trial Registration: ClinicalTrials.gov identifier: NCT03039192
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
See letter by Olie et al
ABSTRACT
Objective: To compare esketamine to placebo, each in addition to standard-of-care treatment, for rapidly reducing major depressive disorder symptoms, including suicidal ideation.
Methods: This phase 3, double-blind, multicenter study (ASPIRE I), conducted between June 2017 and December 2018, enrolled 226 adults having major depressive disorder based on Diagnostic and Statistical Manual of Mental Disorders fifth edition (DSM-5) criteria, active suicidal ideation with intent, and need for psychiatric hospitalization. Patients were randomized 1:1 to esketamine 84 mg or placebo nasal spray twice-weekly for 4 weeks, each with comprehensive standard-of-care treatment (initial psychiatric hospitalization and newly initiated or optimized oral antidepressant[s] therapy). Change from baseline to 24 hours post-first dose in Montgomery-Asberg Depression Rating Scale (MADRS) total score (primary endpoint) was analyzed using analysis of covariance (ANCOVA), and change in Clinical Global Impression of Severity of Suicidality Revised version (CGI-SS-r; key secondary endpoint) score was analyzed using ANCOVA on ranks with treatment difference estimated using the Hodges-Lehmann estimate.
Results: Greater improvement in MADRS total score was observed with esketamine + standard-of-care versus placebo + standard-of-care at 24 hours (least-squares mean difference [SE]: −3.8 [1.39]; 95% CI, −6.56 to −1.09; 2-sided P = .006), as well as at earlier (4 hours) and later time points during 4-week double-blind treatment. The difference between groups in the severity of suicidality was not statistically significant (median of treatment difference [95% CI]: 0.0 [−1.00 to 0.00]; 2-sided P = .107). The most common adverse events among esketamine-treated patients were dizziness, dissociation, headache, nausea, and somnolence.
Conclusions: These findings demonstrate rapid and robust efficacy of esketamine nasal spray in reducing depressive symptoms in severely ill patients with major depressive disorder who have active suicidal ideation with intent.
Trial Registration: ClinicalTrials.gov identifier: NCT03039192
J Clin Psychiatry 2020;81(3):19m13191
To cite: Fu D-J, Ionescu DF, Li X, et al. Esketamine nasal spray for rapid reduction of major depressive disorder symptoms in patients who have active suicidal ideation with intent: double-blind, randomized study (ASPIRE I). J Clin Psychiatry. 2020;81(3):19m13191.
To share: https://doi.org/10.4088/JCP.19m13191
© Copyright 2020 Physicians Postgraduate Press, Inc.
aNeuroscience Clinical Development, Janssen Research & Development, LLC, Titusville, New Jersey
bNeuroscience Clinical Development, Janssen Research & Development, LLC, San Diego, California
cDepartment of Quantitative Sciences, Janssen Research & Development, LLC, Titusville, New Jersey
dDepartment of Psychiatry, Yale School of Medicine, New Haven, Connecticut
*Corresponding author: Dong-Jing Fu, MD, PhD, Director, Neuroscience Clinical Development, Janssen Research and Development, LLC, 1125 Trenton-Harbourton Rd, Titusville, NJ 08560 ([email protected]).
Depression is the leading cause of disability worldwide and a major contributor to the overall global burden of disease.1 Major depressive disorder (MDD) is the psychiatric diagnosis most commonly associated with suicide.2,3 The reported prevalence of suicidal ideation in adult patients with MDD is as high as 60%, and the lifetime incidence of attempted suicide in this population ranges between 10% and 20%.4,5 Further, the lifetime risk of completed suicide has been estimated to be 3.4% in this population.6
Suicidal ideation is a major risk factor for suicide in patients with depression.7,8 The time between the onset of suicidal ideation and suicide attempt is often very short,9 highlighting the need for immediate intervention. Patients with MDD who have active suicidal ideation with intent constitute a psychiatric emergency. These patients are often hospitalized to protect them from self-harm, although the benefits of hospitalization are often temporary. Moreover, while standard antidepressants effectively treat depressive symptomatology, including suicidal ideation,10 they require 4-6 weeks to exert their full effect,11,12 limiting their utility in crisis situations. Currently, there is no approved medication for emergency treatment of patients with depression who have active suicidal ideation with intent.12,13
Esketamine nasal spray was recently approved in the United States and European Union for treating treatment-resistant depression.14,15 Esketamine (the S-enantiomer of ketamine), an N-methyl-d-aspartate (NMDA) receptor antagonist, is thought to confer antidepressant effects by transiently influencing glutamate transmission, increasing neurotrophic factor release, and stimulating synaptogenesis16 through a primary mechanism that is distinct from that of conventional monoaminergic antidepressants.
Four small trials17-20 including patients with MDD suggested that ketamine, administered intravenously, may rapidly decrease suicidal ideation. Further, in a phase 2 double-blind, proof-of-concept study,21 our research group reported that esketamine nasal spray compared with placebo nasal spray, given in addition to comprehensive standard-of-care treatment, resulted in statistically significant and clinically meaningful reduction in depressive symptoms at 4 and 24 hours after the first dose among depressed patients at imminent risk for suicide. The first phase 3 program consisting of 2 identically designed, fully powered global studies (ASPIRE I and ASPIRE II) was undertaken to confirm the antidepressant efficacy of esketamine in this population. The results of ASPIRE I are reported herein.
METHODS
Ethical Practices
Independent Review Boards and Ethics Committees (see Supplementary Appendix 1) approved the study protocol and amendments. The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, consistent with Good Clinical Practices and applicable regulatory requirements. All patients provided written informed consent before participation. The study is registered at https://clinicaltrials.gov/ct2/show/NCT03039192.
Study Population
The study enrolled adults (18-64 years) with a diagnosis of MDD without psychotic features according to Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5),22 and confirmed by the Mini-International Neuropsychiatric Interview (MINI).23 Candidates were screened shortly after presenting to an emergency department or inpatient psychiatric unit. Eligibility criteria required that patients respond affirmatively to MINI questions B3 (“Think about suicide [killing yourself]?”) and B10 (“Intend to act on thoughts of killing yourself in the past 24 hours?”) within 24 hours of randomization, be in clinical need of acute psychiatric hospitalization due to imminent suicide risk, and have a Montgomery-Asberg Depression Rating Scale (MADRS)24 total score > 28 predose on day 1. Patients must have voluntarily agreed to comprehensive standard-of-care treatment, including initial hospitalization and initiation or optimization of a non-investigational antidepressant(s) treatment for at least the duration of double-blind treatment.
Certain psychiatric comorbidities were exclusionary (eg, current DSM-5 diagnosis of bipolar disorder, obsessive-compulsive disorder, antisocial personality disorder, borderline personality disorder), as were moderate-to-severe DSM-5 substance or alcohol use disorder within 6 months prior to screening, current or prior DSM-5 diagnosis of psychotic disorder, and positive urine test result(s) for phencyclidine, cocaine, or amphetamines. A complete list of inclusion and exclusion criteria is presented in Supplementary Appendix 2.
Study Design
This double-blind, randomized, placebo-controlled, multicenter study was conducted from June 2017 to December 2018 at 51 study sites in the United States, Europe, Asia, and South Africa.
The study consisted of a 24- to 48-hour screening period to assess patients’ eligibility, followed by 4-week double-blind treatment (days 1-25) given in the context of comprehensive standard-of-care, and then 9-week posttreatment follow-up (days 26-90). Patients were initially hospitalized in a psychiatric unit for a recommended 5 days, with shorter or longer hospitalizations permitted if clinically warranted per local standard practice.
Eligible patients were randomized (1:1), based on a computer-generated randomization schedule, to 84 mg esketamine nasal spray (referred to as esketamine hereafter) or matching placebo nasal spray (referred to as placebo hereafter), administered twice weekly. Randomization was balanced using randomly permuted blocks and stratified by study center and type of standard-of-care antidepressant (ie, monotherapy or antidepressant plus augmentation therapy) determined by the investigator.
Study Drug and Standard-of-Care Antidepressant Therapy
Intranasal study drugs were provided in disposable nasal spray devices with identical appearance and packaging. Each device contained 200 μL of solution and delivered 2 sprays of either esketamine (total of 28 mg of esketamine base) or placebo. The placebo solution contained a bittering agent to simulate the taste of esketamine solution, and the same number of devices (3) were administered to all patients at all sessions.
Patients self-administered study drug, under the supervision of a site staff member, twice weekly for 4 weeks. After day 1, a single dose reduction of esketamine (or placebo) from 84 mg to 56 mg was permitted for intolerance, with the 56-mg dose continued thereafter.
Standard-of-care oral antidepressant(s) treatment (either monotherapy or antidepressant + augmentation therapy) was initiated or optimized at the time of randomization on day 1 by the investigator based on clinical judgment and practice guidelines. Augmenting agents could consist of a second antidepressant, an atypical antipsychotic, or a mood stabilizer.
Dose titration/adjustments of standard-of-care antidepressant(s) occurred during the first 2 weeks of double-blind treatment, after which doses were to remain stable. During the follow-up phase, patients were treated with standard-of-care antidepressant(s) managed per clinical judgment.
Efficacy Assessments
- Currently, there is no approved medication for emergency treatment of patients with depression who have active suicidal ideation with intent.
- Esketamine nasal spray rapidly reduced depressive symptoms in adult patients with major depressive disorder who had moderate to severe depression and suicidal ideation with intent.
Depressive symptom severity was assessed using the Structured Interview Guide for MADRS24 on day 1 (predose and 4 hours postdose), day 2 (~ 24 hours postdose), all subsequent visits (predose), at 4 hours postdose on day 25 during the double-blind phase, and at all visits during the follow-up phase (twice-weekly through day 39, weekly through day 53, and every other week through day 90). For the 4-hour version of the MADRS, the Reduced Sleep item was not assessed, but the scores for the Reduced Sleep item recorded predose on the same day were carried forward and included in the total score.
Efficacy related to suicidal ideation and behavior was assessed using the Suicide Ideation and Behavior Assessment Tool (SIBAT; see Supplementary Figure 1), a computerized instrument,25 on all visit days during the double-blind (predose; 4 hours postdose on day 1) and follow-up phases. The SIBAT contains both patient- and clinician-reported modules, which include assessments of Clinical Global Impression of Severity of Suicidality Revised version (CGI-SS-r; rated from 0 [normal, not at all suicidal] to 6 [among the most extremely suicidal patients]), Clinical Global Impression of Imminent Suicide Risk (CGI-SR-I), and, clinician-rated and patient-reported Frequency of Suicidal Thinking (FoST).
Click figure to enlarge
Safety Assessments
Adverse events were monitored throughout the study. Vital signs were assessed and the Clinician Administered Dissociative States Scale (CADSS)26 and Modified Observer’s Assessment of Alertness Sedation (MOAA/S)27 were administered at all dosing visits. The SIBAT was also utilized as a safety outcome.
To maintain the study blind, efficacy and safety assessments were performed by different raters who were trained and certified.
Statistical Methods
All randomized patients who received at least 1 dose of double-blind study medication were included in the safety analysis set. The full efficacy analysis set included all patients in the safety analysis set who had both a baseline and ≥ 1 postbaseline evaluation with the MADRS or CGI-SS-r. The follow-up analysis set included all patients who completed the double-blind treatment phase and either entered the follow-up phase or provided adverse event data after the double-blind treatment phase.
Sample Size Determination
Sample size was calculated based on an effect size of 0.45 for the change in MADRS total score between esketamine and placebo, a 2-sided significance level of .050, and a dropout rate of 5% at 24 hours. Approximately 112 patients were to be randomized to each treatment group to achieve 90% power.
Efficacy Endpoints and Analyses
Statistical analysis tests were conducted at a 2-sided .050 significance level. A fixed sequence approach was applied to adjust for multiplicity and to control type I error for the primary and key secondary efficacy endpoints (ie, secondary efficacy endpoint was tested only after rejecting the null hypothesis for the primary endpoint).
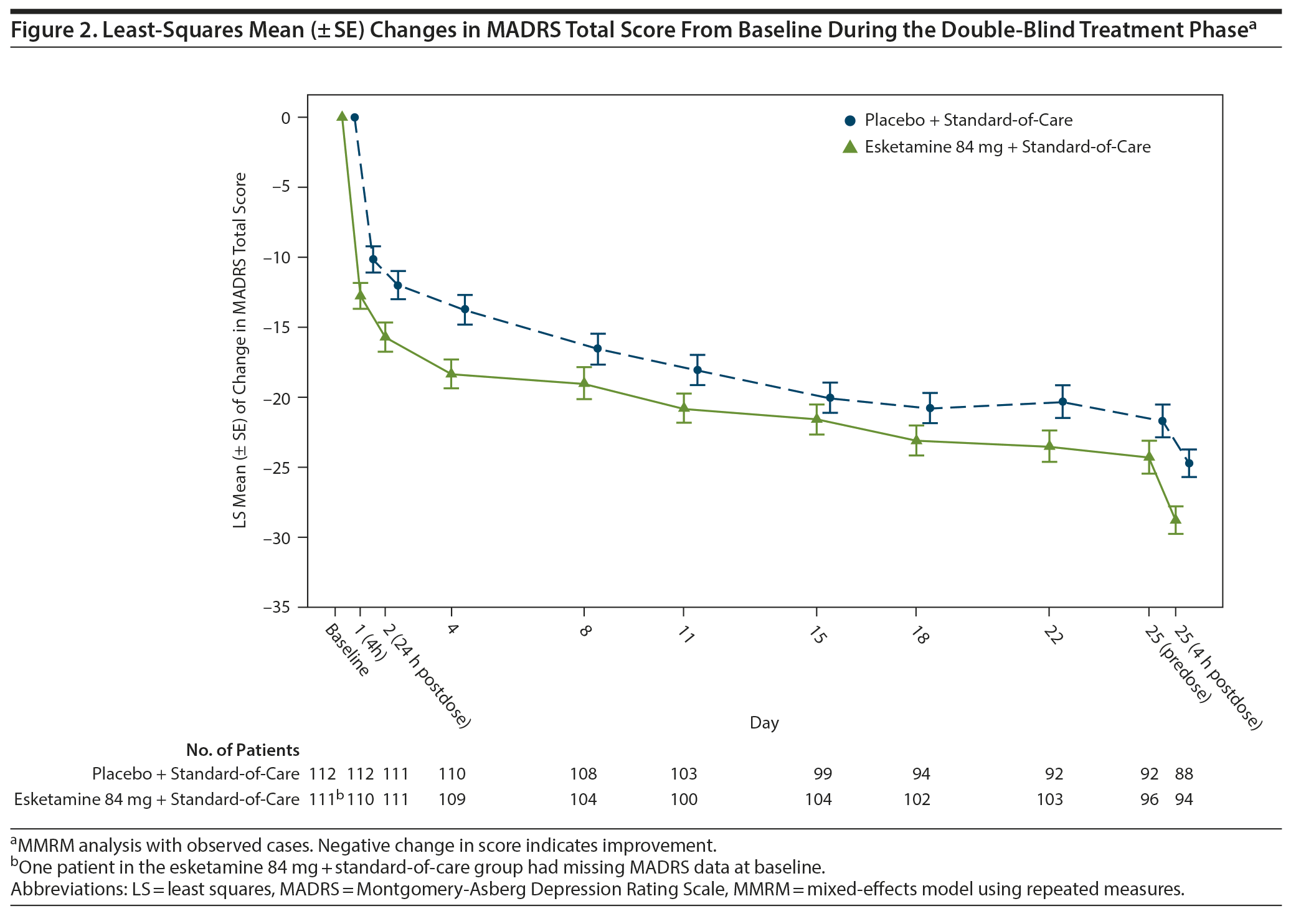
The primary efficacy endpoint—change in MADRS total score from baseline (day 1, predose) to 24 hours post-first dose (day 2)—was analyzed using the analysis of covariance (ANCOVA) model with treatment (placebo or esketamine 84 mg), standard-of-care antidepressant as randomized (monotherapy or antidepressant + augmentation therapy), and analysis center as factors and baseline MADRS total score as a continuous covariate. Missing day 2 MADRS total score was carried forward from 4 hours for 1 patient. A mixed model for repeated measures (MMRM) was used to explore the course of treatment effect over time for the MADRS total score during the double-blind and follow-up phases.
The key secondary endpoint—change in CGI-SS-r score from baseline to 24 hours after the first dose—was analyzed using an ANCOVA model on the ranks of change with the same factors (noted in the previous paragraph) and unranked baseline score as a covariate. The median of treatment difference was estimated using the Hodges-Lehmann estimate.
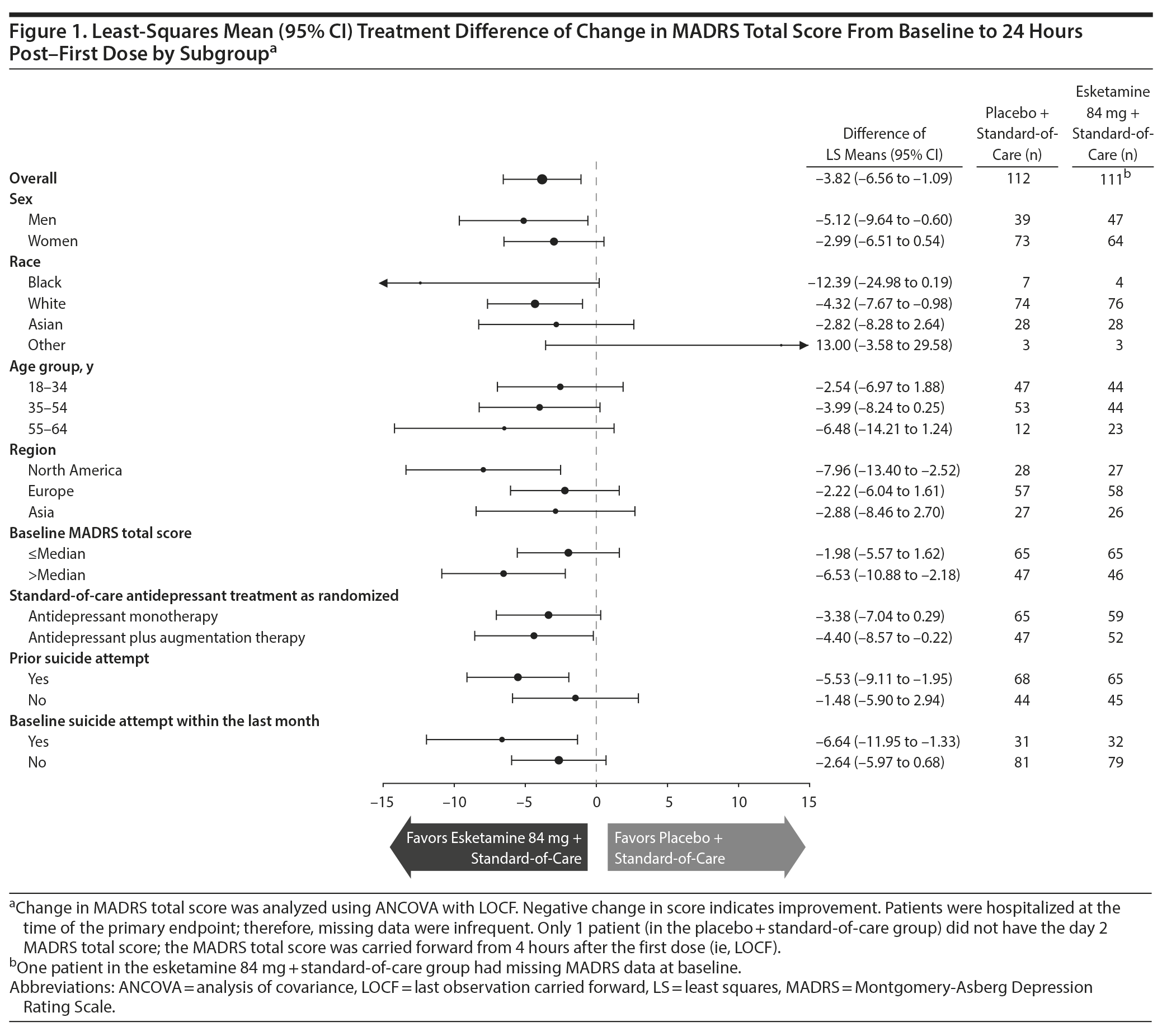
Prespecified subgroup analyses (shown in Figure 1 and Supplementary Figure 5) were conducted according to an ANCOVA model for the primary endpoint and key secondary endpoint (using unranked data), respectively.
Data for patients in remission (MADRS score ≤ 12) over time were summarized, and estimates of the treatment difference in proportions and 95% confidence intervals (CIs) were provided. Differences in least-squares means and 95% CIs were provided for other suicidality indices (CGI-SR-I, clinician- and patient-rated FoST, MADRS suicide item) based on ANCOVA modeling similar to that described for the primary analysis.
Frequency distributions or descriptive statistics were provided for adverse events, vital signs, and scores for clinician-reported outcomes (MOAA/S, CADSS).
RESULTS
Patients and Treatment
A total of 226 patients were randomized (114 and 112 to esketamine + standard-of-care and placebo + standard-of-care, respectively) (Supplementary Figure 2). Of the patients randomized to esketamine + standard-of-care, 1 was excluded from the safety analysis set and the full efficacy analysis set because the patient did not receive any dose of study drug. Another patient randomized to esketamine + standard-of-care was excluded from the full efficacy analysis set because the patient discontinued after the first dose of study agent on day 1 and did not provide any efficacy data after baseline (day 1, predose). Most randomized patients (esketamine + standard-of-care: 102/114 [89.5%]; placebo + standard-of-care: 93/112 [83.0%]) completed the double-blind treatment phase; 192 entered the follow-up phase, with 164 completing the day 90 follow-up visit.
Click figure to enlarge
The treatment groups were similar with respect to demographic and baseline clinical characteristics (Table 1), standard-of-care antidepressant use, and concomitant use of benzodiazepines. At baseline, mean MADRS total score was 41.1. The majority (60.1%) of patients reported a prior suicide attempt, 28.1% within the last month. The investigator rated most patients (88.8%) to be moderately to extremely suicidal, as measured by the CGI-SS-r. The most frequently reported standard-of-care antidepressant therapies were venlafaxine (24.9%), escitalopram (16.0%), duloxetine (15.6%), mirtazapine (15.6%), and quetiapine (14.2%). Approximately three-fourths of patients in the safety analysis dataset received ≥ 1 concomitant benzodiazepine during the double-blind treatment phase.
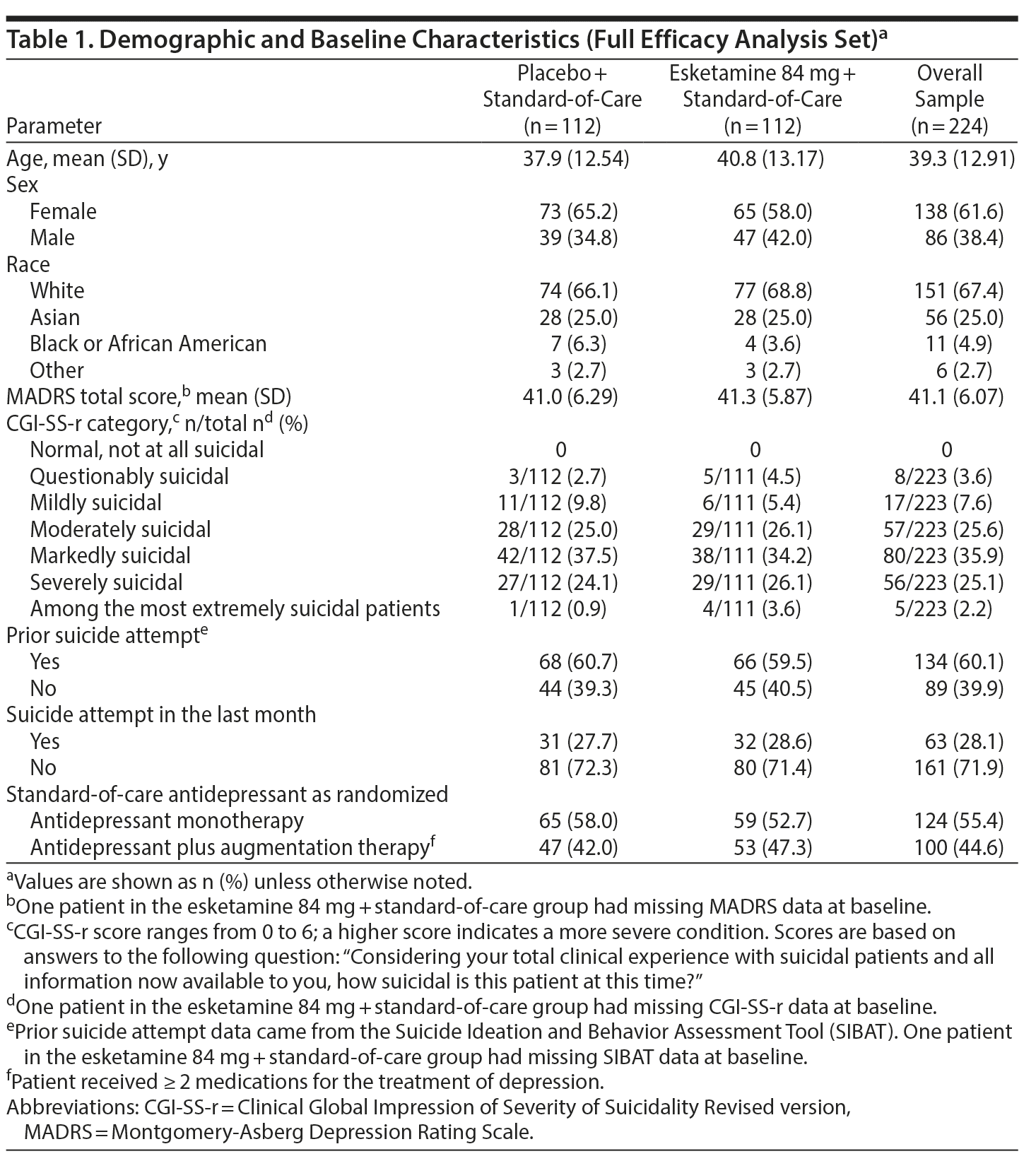
Click figure to enlarge
Efficacy Results
Symptoms of depression. In analysis of the primary endpoint, MADRS total score decreased (improved) from baseline to 24 hours after the first dose (day 2) in both the esketamine + standard-of-care (mean [SD]: -16.4 [11.95]) and placebo + standard-of-care groups (-12.8 [10.73]), with significantly greater improvement with esketamine (least-squares mean difference [SE]: -3.8 [1.39]; 95% CI, −6.56 to −1.09; 2-sided P = .006). The mean between-group difference [95% CI] in MADRS total score at 24 hours favored esketamine in most subgroups (Figure 1), notably so among patients with prior suicide attempt (−5.53 [−9.11 to −1.95]) and patients with more severe depressive symptoms (ie, MADRS total score > median) (−6.53 [−10.88 to −2.18]).
The treatment effect of esketamine on depressive symptoms was observed starting at 4 hours after the first dose. Patients in both groups continued to improve over the double-blind treatment phase; the difference between treatment groups generally remained over time through day 25 (Figure 2). MADRS total scores were similar between groups and remained low throughout follow-up (Supplementary Figure 3).
Click figure to enlarge
The percentages of patients who achieved remission (MADRS total score ≤ 12) are presented in Supplementary Figure 4. The treatment difference (95% CI) was 9.8% (0.87 to 18.77) 24 hours post-first dose and 16.1% (3.20 to 28.94) on day 25, 4 hours postdose.
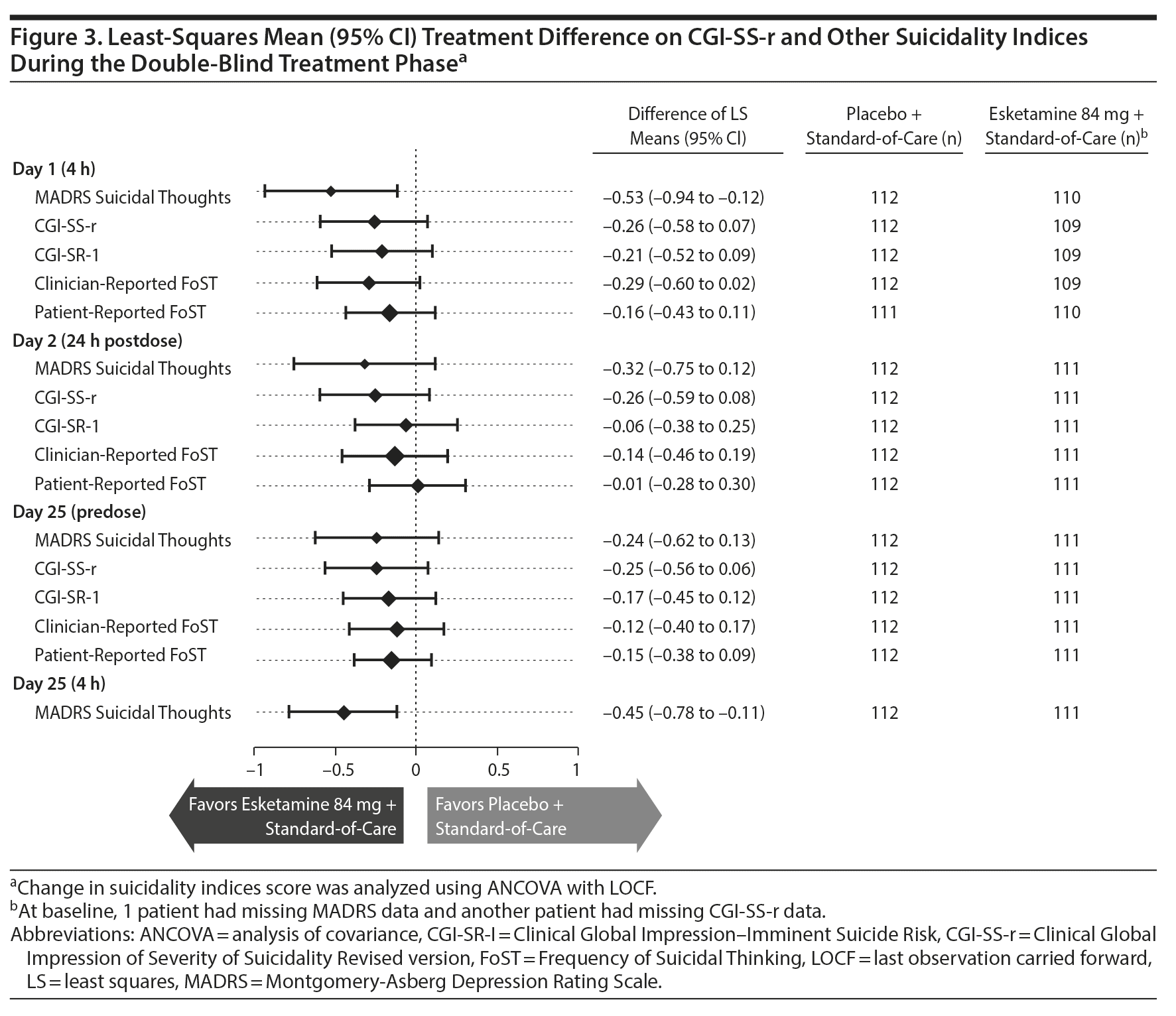
Severity of suicidality. At the 24-hour endpoint, patients in both treatment groups experienced improvement in the severity of their suicidality as measured by CGI-SS-r, though there was no statistically significant difference between treatment groups (2-sided P = .107). The Hodges-Lehmann estimate of the treatment difference (95% CI) was 0.0 (−1.00 to 0.00).
The estimated differences (95% CI) between treatment groups at 24 hours post-first dose with esketamine for the change in CGI-SS-r score were -0.40 (−0.84 to 0.04) for patients with a history of prior suicide attempt and −0.60 (−1.14 to −0.06) for patients with more severe depressive symptoms (Supplementary Figure 5).
Improvement in severity of suicidality was also observed in both treatment groups at the end of double-blind treatment (Supplementary Figure 6). Results for other indices of suicidality are presented in Figure 3.
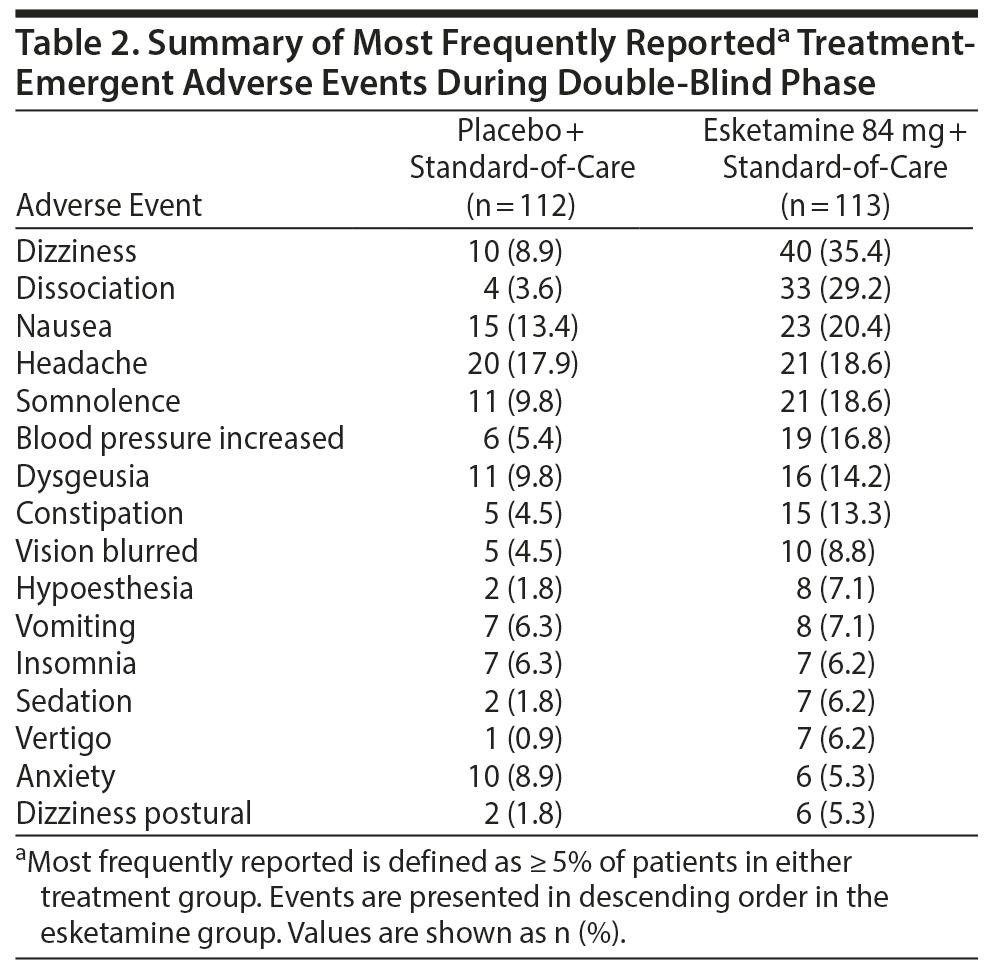
Safety Results
The adverse events most frequently reported during the double-blind treatment phase are shown in Table 2 (those most frequently reported during the follow-up phase, in Supplementary Table 1). Most events in the esketamine + standard-of-care (91.0%) and placebo + standard-of-care (70.3%) groups occurred on intranasal dosing days, and most of these (94.9%; and 85.7%, respectively) resolved on the same day. Twenty-one patients (18.6%) in the esketamine + standard-of-care group had a dose reduction to 56 mg due to intolerance, primarily on second dosing.
Click figure to enlarge
No deaths were reported during double-blind treatment. Serious adverse events are presented in Supplementary Table 2 and Supplementary Table 3, and events leading to discontinuation of study drug are summarized in Supplementary Appendix 3.
All depression- and suicide-related adverse events reported in the double-blind treatment phase were considered by the study site investigator as unrelated to esketamine. Suicide attempt was reported for 1 patient in each treatment group during the double-blind phase. Suicide-related serious adverse events during the follow-up phase—including 3 suicide attempts and 1 completed suicide among patients who had prior esketamine treatment and 2 suicide attempts among patients who had prior placebo treatment—were dispersed, without pattern or signal of rebound. The 1 patient who died by suicide in the follow-up phase, 3 days after receiving her last esketamine dose, had a history of 5 prior suicide attempts, the most recent in the month prior to randomization. All patients who attempted suicide during the study had also made an attempt within the month prior to randomization.
The results of assessments of CADSS and blood pressure are provided in Supplementary Figure 7 and Supplementary Figure 8, respectively. More patients in the esketamine + standard-of-care group (13/113 [11.5%]) had an MOAA/S score ≤ 3 (indicating moderate or greater sedation) at any time during the double-blind phase, versus placebo + standard-of-care (1/112 [0.9%]), and none of these patients required medical intervention.
DISCUSSION
This study is pivotal to the first global registration program of patients with MDD and active suicidal ideation with intent, a population typically excluded from antidepressant treatment trials28 and for whom no approved pharmacologic treatment exists. The results of this phase 3 study demonstrate that esketamine nasal spray rapidly reduces depressive symptoms in this very ill, vulnerable population. The clinical benefit of esketamine is notable given the large nonspecific benefits afforded by the background of comprehensive standard-of-care,29 consisting of initial inpatient psychiatric hospitalization and newly initiated or optimized antidepressant therapy. Specifically, improvement in MADRS total score was greater with esketamine than placebo starting at 4 hours post-first dose and continuing until the end of double-blind treatment, when the newly initiated or optimized antidepressant therapy had sufficient time to exert its effect.
In addition to the observed clear benefit of esketamine on depressive symptoms, patients in both the esketamine + standard-of-care and placebo + standard-of-care groups experienced rapid reduction in the severity of their suicidality, as measured by CGI-SS-r at 24 hours; however, the difference between treatment groups was not statistically significant. This may be due to the substantial impact of inpatient psychiatric hospitalization in diffusing the acute suicidal crisis. Further, comprehensive standard-of-care was enhanced by twice-weekly study visits with extensive clinical contact and permitted benzodiazepine use, all of which may have contributed to the rapid reduction of suicidality in both treatment groups.
Intravenous ketamine has been reported to rapidly reduce suicidal ideation, although most of these trials did not specifically select patients with active suicidal ideation and at imminent risk for suicide,30 as required by our study. Although we also observed rapid treatment effect (4 hours post-first dose) with esketamine on measures of suicidality in a phase 2 proof-of-concept study with similar design and patient population,21 those results were not confirmed in this phase 3 study. This lack of confirmation may be due to increased heterogeneity of the patient population and standard-of-care associated with a large global study.
Adverse events observed in this study are consistent with the established safety profile of esketamine nasal spray.14,15 Sadly, 1 study patient, treated with esketamine during the double-blind phase, died by suicide during the follow-up phase. The patient had a history of multiple prior suicide attempts, including one within the month prior to randomization. This was the only completed suicide across the entire clinical development program in over 500 patients who had active suicidal ideation with intent. This low number of completed suicides very likely reflects the comprehensive clinical care and close follow-up patients received during the study.
Study Limitations
Conducting clinical trials in a population of patients with MDD who have active suicidal ideation and intent presents unique methodological challenges, including parsing the benefits of hospitalization, as well as the care, attention, and expectancy bias that accompanies participation in research.31 In the study of this high-risk patient population, these methodological challenges may be inevitable to ensure ethical practice and patient safety. Also noteworthy are potential regional differences in the standard-of-care treatment provided in this global study. As esketamine has known transient sedative and dissociative effects, patients themselves may have been unblinded. To mask the bitter taste of esketamine, a bittering agent was added to the placebo nasal spray. To ensure that efficacy raters were not unblinded, different raters were used to perform efficacy and safety assessments.
CONCLUSIONS
Taken together, our findings suggest esketamine nasal spray may address the unmet need for a rapid-acting antidepressant in patients with MDD and active suicidal ideation with intent, for which there is no approved pharmacologic treatment.
Submitted: December 2, 2019; accepted April 14, 2020.
Published online: May 12, 2020.
Author contributions: Drs Fu, Canuso, Li, Lim, and Ionescu and Ms Lane participated in study design, data collection, data analysis and interpretation, and writing and review of the manuscript. Drs Hough, Drevets, Manji, and Sanacora participated in study design, data analysis and interpretation, and review of the manuscript. Drs Li and Lim and Ms Lane participated in the statistical design. All authors meet ICMJE criteria, and all those who fulfilled those criteria are listed as authors.
Potential conflicts of interest: Drs Fu, Ionescu, Li, Lim, Hough, Drevets and Canuso and Ms Lane are employees of Janssen Research & Development, LLC. Dr Manji is an employee of Janssen Research & Development, LLC and is an inventor on patents that are directed to this technology, are assigned to Icahn School of Medicine at Mount Sinai, Yale University, and the National Institutes of Health (NIH), and are exclusively licensed to Janssen; however, he does not receive any direct financial benefit therefrom. Dr Sanacora has received consulting fees from Allergan, Alkermes, Axsome Therapeutics, Boehringer Ingelheim, Biohaven, Clexio Biosciences, Epiodyne, Intra-Cellular Therapies, Janssen, Merck, Navitor, NeruoRx, Novartis, Noven, Otsuka, Perception Neuroscience, Praxis Therapeutics, Sage, Taisho, Valeant, and Vistagen Therapeutics over the last 24 months. He has also received additional research contracts from Johnson & Johnson and Merck over the last 36 months. Free medication was provided to Dr Sanacora for an NIH-sponsored study by Sanofi-Aventis. In addition, he holds shares in BioHaven Pharmaceuticals Holding Company and is a co-inventor on the patent “Glutamate agents in the treatment of mental disorders” (Patent number: 8778979) and a US Provisional Patent Application No. 047162-7177P1 (00754) filed on August 20, 2018, by Yale University Office of Cooperative Research OCR 7451 US01. Dr Sanacora’s employer, Yale University, has a financial relationship with Janssen Pharmaceuticals and may in the future receive financial benefits from this relationship. The University has put multiple measures in place to mitigate this institutional conflict of interest. Questions about the details of these measures should be directed to Yale University’s Conflict of Interest office.
Funding/support: This study was funded by Janssen Research & Development, LLC, Titusville, NJ.
Role of the sponsor: Employees of Janssen Research & Development, LLC, as noted under Author Contributions, were involved in design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Disclaimer: All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Previous presentations: Poster presented at the 32nd ECNP Congress; September 7-10, 2019; Copenhagen, Denmark ▪ US Psych Congress 2019; October 3-6, 2019; San Diego, California, ▪ Suicide Research Summit 2019 (IASR/AFSP); October 27-30, 2019; Miami, Florida.
Acknowledgments: Anna Duca, MSN; Valerie Joanny, MS; Susan Katz, MS; and Christine Pinter, MS (Janssen Research & Development, LLC) provided operational support for study execution. Sandra Norris, PharmD, of the Norris Communications Group LLC, supported by Janssen Research & Development, LLC, provided medical writing assistance. Ellen Baum, PhD (Janssen Global Services, LLC) provided additional editorial support and was not compensated.
We thank the study patients for their participation in this study. The following principal investigators participated in the study. Investigators received fair market value compensation for their participation in this study without inducement for authorship or other benefit.
BULGARIA: Valentin Akabaliev, Dancho Dilkov, Temenuzhka Mateva, Mitko Mitev, Veselin Palazov; ESTONIA: Innar Toru, Ulle Vohma; GERMANY: Stephanie Krüger, Claus Normann, Andreas Reif; HUNGARY: Sandor Fekete, Gábor Feller, Tamás Kurimay, János Réthelyi, Zsuzsanna Somlai; KOREA: Yong Min Ahn, Changsu Han, Jinpyo Hong, Jae-min Kim, Jong-woo Paik; MALAYSIA: Akramul Zikri Abdul Malek, Ahmad Hatim Sulaiman, Kit Chan Wong; SOUTH AFRICA: Daniel Niehaus, Juan Schrönen; SPAIN: Angela Ibanez, Sonia Bustamante Madariaga, Salvador Sarro Maluquer, Salvador Ros Montalbán, Francisco Monta×±és Rada, Josep Antoni Ramos Quiroga, Patricio Molero Santos, Eduard Vieta; TAIWAN: Po-chung Ju, Hsin-chien Lee, Cheng-ta Li, Chin-bin Yeh; UNITED STATES: Alabama: Richard Shelton; California: David Walling; Connecticut: Gerard Sanacora; Illinois: Constantin Abuzatoaie, John Zajecka; Kentucky: Rifaat S. El-Mallakh; Maryland: Scott Aaronson, Robert Litman; New York: Steven Dubovsky, Michael Grunebaum; North Carolina: James Barker; Ohio: Subhdeep Virk; South Carolina: Robert Malcolm; Texas: Manish Jha.
Additional information: The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. Requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
Supplementary material: See accompanying pages.
REFERENCES
1.World Health Organization. Depression. WHO website: https://www.who.int/news-room/fact-sheets/detail/depression/. March 2018. Accessed October 24, 2019.
2.Harris EC, Barraclough B. Suicide as an outcome for mental disorders. a meta-analysis. Br J Psychiatry. 1997;170(3):205-228. PubMed CrossRef
3.Kessler RC, Berglund P, Borges G, et al. Trends in suicide ideation, plans, gestures, and attempts in the United States, 1990-1992 to 2001-2003. JAMA. 2005;293(20):2487-2495. PubMed CrossRef
4.Hasin DS, Sarvet AL, Meyers JL, et al. Epidemiology of adult DSM-5 major depressive disorder and its specifiers in the United States. JAMA Psychiatry. 2018;75(4):336-346. PubMed CrossRef
5.Holma KM, Melartin TK, Haukka J, et al. Incidence and predictors of suicide attempts in DSM-IV major depressive disorder: a five-year prospective study. Am J Psychiatry. 2010;167(7):801-808. PubMed CrossRef
6.Blair-West GW, Cantor CH, Mellsop GW, et al. Lifetime suicide risk in major depression: sex and age determinants. J Affect Disord. 1999;55(2-3):171-178. PubMed CrossRef
7.Bickley H, Hunt IM, Windfuhr K, et al. Suicide within two weeks of discharge from psychiatric inpatient care: a case-control study. Psychiatr Serv. 2013;64(7):653-659. PubMed CrossRef
8.McAuliffe CM. Suicidal ideation as an articulation of intent: a focus for suicide prevention? Arch Suicide Res. 2002;6(4):325-338. CrossRef
9.Deisenhammer EA, Ing CM, Strauss R, et al. The duration of the suicidal process: how much time is left for intervention between consideration and accomplishment of a suicide attempt? J Clin Psychiatry. 2009;70(1):19-24. PubMed CrossRef
10.Montgomery SA, Dunner DL, Dunbar GC. Reduction of suicidal thoughts with paroxetine in comparison with reference antidepressants and placebo. Eur Neuropsychopharmacol. 1995;5(1):5-13. PubMed CrossRef
11.Simon GE, Savarino J. Suicide attempts among patients starting depression treatment with medications or psychotherapy. Am J Psychiatry. 2007;164(7):1029-1034. PubMed CrossRef
12.Wasserman D, Rihmer Z, Rujescu D, et al; European Psychiatric Association. The European Psychiatric Association (EPA) guidance on suicide treatment and prevention. Eur Psychiatry. 2012;27(2):129-141. PubMed CrossRef
13.Valenstein M, Kim HM, Ganoczy D, et al. Higher-risk periods for suicide among VA patients receiving depression treatment: prioritizing suicide prevention efforts. J Affect Disord. 2009;112(1-3):50-58. PubMed CrossRef
14.Spravato (esketamine) nasal spray [package insert]. Janssen Pharmaceuticals Companies, Titusville, NJ; 2019.
15.Spravato (esketamine) nasal spray EMA Summary of Product Characteristics. Europen Medicines Agency website. https://www.ema.europa.eu/en/documents/product-information/spravato-epar-product-information_en.pdf.
16.Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338(6103):68-72. PubMed CrossRef
17.Ballard ED, Luckenbaugh DA, Richards EM, et al. Assessing measures of suicidal ideation in clinical trials with a rapid-acting antidepressant. J Psychiatr Res. 2015;68:68-73. PubMed CrossRef
18.Ionescu DF, Swee MB, Pavone KJ, et al. Rapid and sustained reductions in current suicidal ideation following repeated doses of intravenous ketamine: secondary analysis of an open-label study. J Clin Psychiatry. 2016;77(6):e719-e725. PubMed CrossRef
19.Murrough JW, Soleimani L, DeWilde KE, et al. Ketamine for rapid reduction of suicidal ideation: a randomized controlled trial. Psychol Med. 2015;45(16):3571-3580. PubMed CrossRef
20.Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335. PubMed CrossRef
21.Canuso CM, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine for the rapid reduction of symptoms of depression and suicidality in patients at imminent risk for suicide: results of a double-blind, randomized, placebo-controlled study. Am J Psychiatry. 2018;175(7):620-630. PubMed CrossRef
22.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5). Fifth Edition. Washington, DC, American Psychiatric Association; 2013.
23.Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(suppl 20):22-33, quiz 34-57. PubMed
24.Williams JB, Kobak KA. Development and reliability of a structured interview guide for the Montgomery-Asberg Depression Rating Scale (SIGMA). Br J Psychiatry. 2008;192(1):52-58. PubMed CrossRef
25.Alphs L, Fu D-J, Williamson D, et al. Validation and mapping of the Suicidal Ideation and Behavior Assessment Tool (SIBAT). (abstract W88) Neuropsychopharmacology. 2018;43:S427-S428.
26.Bremner JD, Krystal JH, Putnam FW, et al. Measurement of dissociative states with the Clinician-Administered Dissociative States Scale (CADSS). J Trauma Stress. 1998;11(1):125-136. PubMed CrossRef
27.Chernik DA, Gillings D, Laine H, et al. Validity and reliability of the Observer’s Assessment of Alertness/Sedation Scale: study with intravenous midazolam. J Clin Psychopharmacol. 1990;10(4):244-251. PubMed
28.Ballard ED, Snider SL, Nugent AC, et al. Active suicidal ideation during clinical antidepressant trials. Psychiatry Res. 2017;257:303-308. PubMed CrossRef
29.Leuchter AF, Hunter AM, Tartter M, et al. Role of pill-taking, expectation and therapeutic alliance in the placebo response in clinical trials for major depression. Br J Psychiatry. 2014;205(6):443-449. PubMed CrossRef
30.Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158. PubMed CrossRef
31.Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. 2013;170(7):723-733. PubMed CrossRef
Editor’s Note: We encourage authors to submit papers for consideration as a part of our Focus on Suicide section. Please contact Philippe Courtet, MD, PhD, at [email protected].
This PDF is free for all visitors!