Abstract
Background: Onfasprodil (MIJ821) is a highly potent and novel selective NR2B subunit negative allosteric modulator. This phase 2, randomized, placebo-controlled, proof-of-concept study evaluated efficacy and safety of onfasprodil in patients with treatment-resistant major depression (TRD).
Methods: Adults with TRD who did not respond to ≥2 antidepressants were randomized (3:3:3:3:6:4) to receive a 40-minute intravenous infusion of onfasprodil 0.16 mg/kg weekly (n = 11), onfasprodil 0.16 mg/kg biweekly (n = 10), onfasprodil 0.32 mg/kg weekly (n = 10), onfasprodil 0.32 mg/kg biweekly (n = 9), placebo weekly (n=20), or ketamine 0.5 mg/kg weekly (n= 10) for 6 weeks. Primary end point was change from baseline in Montgomery-Asberg Depression Rating Scale (MADRS) score at 24 hours. Secondary end points were change in MADRS score at 48 hours and at final follow-up at 6 weeks. Safety and tolerability were assessed during the study.
Results: Of 70 randomized patients, 53 (75.7%) completed the study. At 24 hours, adjusted mean differences versus placebo for pooled onfasprodil 0.16 mg/kg, 0.32 mg/kg, and ketamine groups were −8.25 (P = .001), −5.71 (P = .019), and −5.67 (P = .046), and at 48 hours, −7.06 (P = .013), −7.37 (P = .013), and −11.02 (P = .019), respectively. At Week 6, adjusted arithmetic mean MADRS difference between ketamine and placebo was −5.24 (80% CI, −10.42 to −0.06; P= .0974). At Week 6, the difference versus placebo on MADRS was −5.78 (P= .0427) for pooled 0.16 mg/kg and −4.24 (P= .1133) for pooled 0.32 mg/kg groups. The commonest treatment-emergent adverse events in the onfasprodil groups were dizziness (14.3%), transient amnesia (14.3%), and somnolence (11.4%). It had overall a good safety profile and was well tolerated.
Conclusion: Onfasprodil appeared to be effective and well-tolerated across all dosing regimens in patients with TRD and demonstrated rapid onset of action (24 hours) with evidence of antidepressant effects to be maintained at Week 6, particularly for the lower-dose group.
Trial Registration: ClinicalTrials.gov identifier: NCT03756129.
J Clin Psychiatry 2025;86(3):23m15246
Author affiliations are listed at the end of this article.
Treatment-resistant depression (TRD) is defined as inadequate response to at least 2 different antidepressants for at least 6 weeks at an adequate dose.1 About one-third of patients with major depressive disorder (MDD) do not respond to antidepressant treatment, and the majority does not maintain a long-term response to standard antidepressants and can be considered treatment-resistant.2 There is a high unmet medical need for effective and well-tolerated rapid-acting antidepressants that can effectively end a depressive episode and prevent future depression.
Ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist, has been shown to be effective in TRD due to its rapid onset, and ability to reduce suicidality, and sustained benefit with repeated dosing,3,4 but it is associated with reversible dissociative and psychotomimetic effects that sometimes can be severe.5 Esketamine (nasal spray), an enantiomer of ketamine approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) for TRD, also shares these risks.
Traxoprodil (CP-101,606), a negative allosteric modulator (NAM) selective for the NR2B subtype of the NMDA receptor, was also effective in patients with TRD, with a magnitude and duration of response comparable to that of ketamine, but without producing a dissociative reaction at the lower dose infusion.6 The development of traxoprodil was halted because of QTc prolongation.7,8
Onfasprodil (MIJ821), a highly potent and selective NMDA NR2B subunit NAM, is expected to exert a rapid antidepressant effect compared to traxoprodil and ketamine; therefore, it is intended to be studied in TRD. Onfasprodil has shown low rates of psychotomimetic side effects in a first-in-human study.9 Hence, onfasprodil could be a potential treatment option for patients with TRD. We evaluated the efficacy and safety of onfasprodil in patients with TRD in a proof-of-concept study.
METHODS
Study Design
This was a phase 2, randomized, double-blind, placebo-controlled, parallel-group, multicenter study (ClinicalTrials.gov identifier NCT03756129) with 6 treatment arms conducted in Spain and with 5 treatment arms in the United States where the ketamine arm was not included. The total study duration was 14 weeks and comprised a screening period (maximum 4 weeks), a 36-day treatment period, and a 5-week follow-up period (Figure 1A).
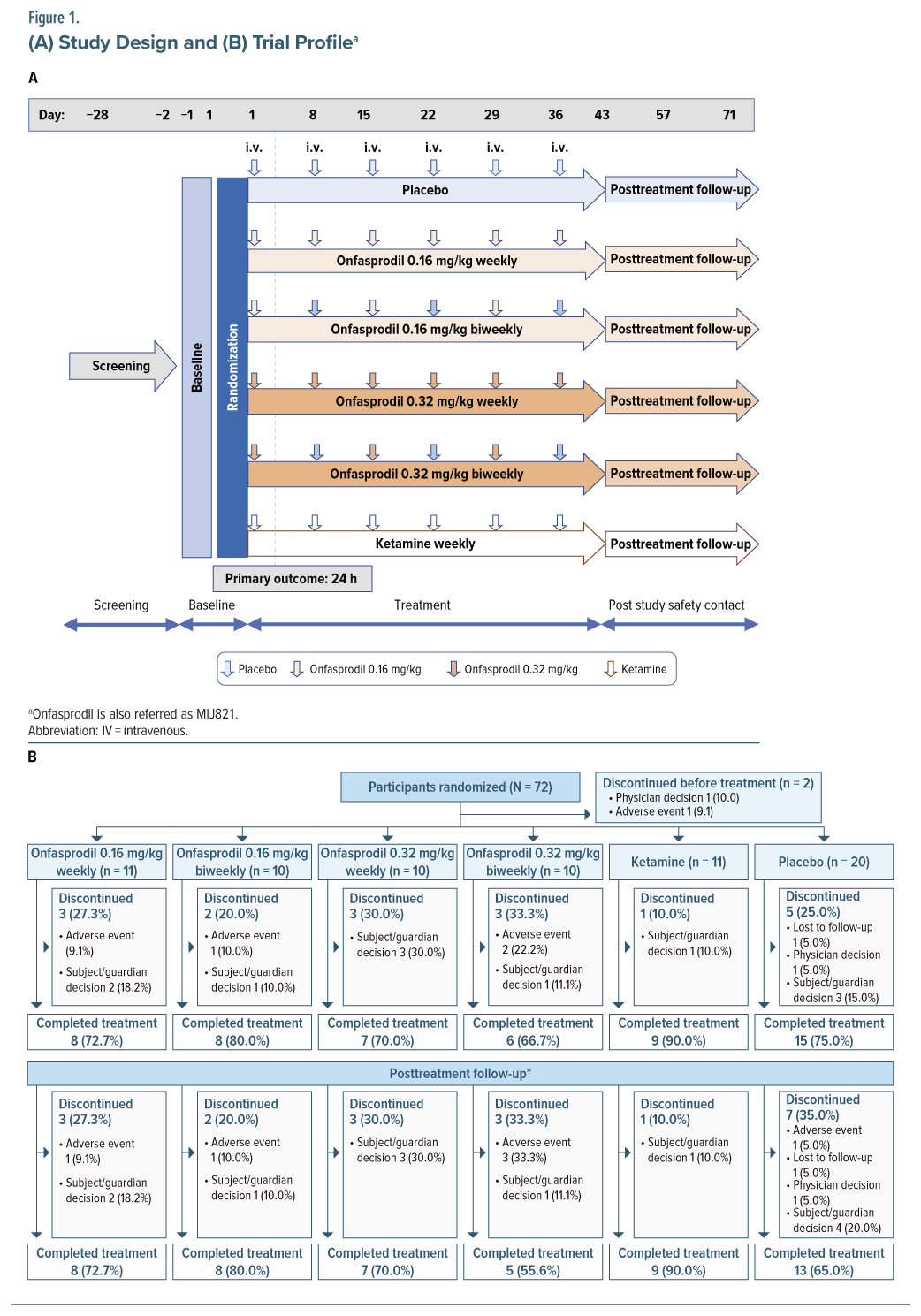
Study Treatment and Randomization
Seventy patients were randomized (3:3:3:3:6:4) to one of the following treatment arms: onfasprodil 0.16 mg/kg 1 infusion per week from Day 1 to Day 36; onfasprodil 0.16 mg/kg 1 infusion biweekly on Day 1, Day 15, and Day 29 and placebo on Day 8, Day 22, and Day 36; onfasprodil 0.32 mg/kg 1 infusion per week from Day 1 to Day 36; onfasprodil 0.32 mg/kg 1 infusion biweekly on Day 1, Day 15, and Day 29 and placebo on Day 8, Day 22, and Day 36; placebo 1 infusion per week from Day 1 to Day 36; and ketamine 0.5 mg/kg, limiting dose at 40 mg/infusion for patients over 80 kg, 1 infusion per week from Day 1 to Day 36 (absence of the ketamine arm in the US). All study treatments were administered via a 40-minute intravenous infusion at the site. Based on preclinical rodent PET studies, both doses (0.16 and 0.32 mg/kg) were projected to give a maximum receptor occupancy greater than 90% of brain NR2B receptors (data on file).
Randomization was implemented using Interactive Response Technology, using a validated system that automated the random assignment of patient numbers to randomization numbers. Randomization was stratified by region, the United States, and European countries.
Study Population
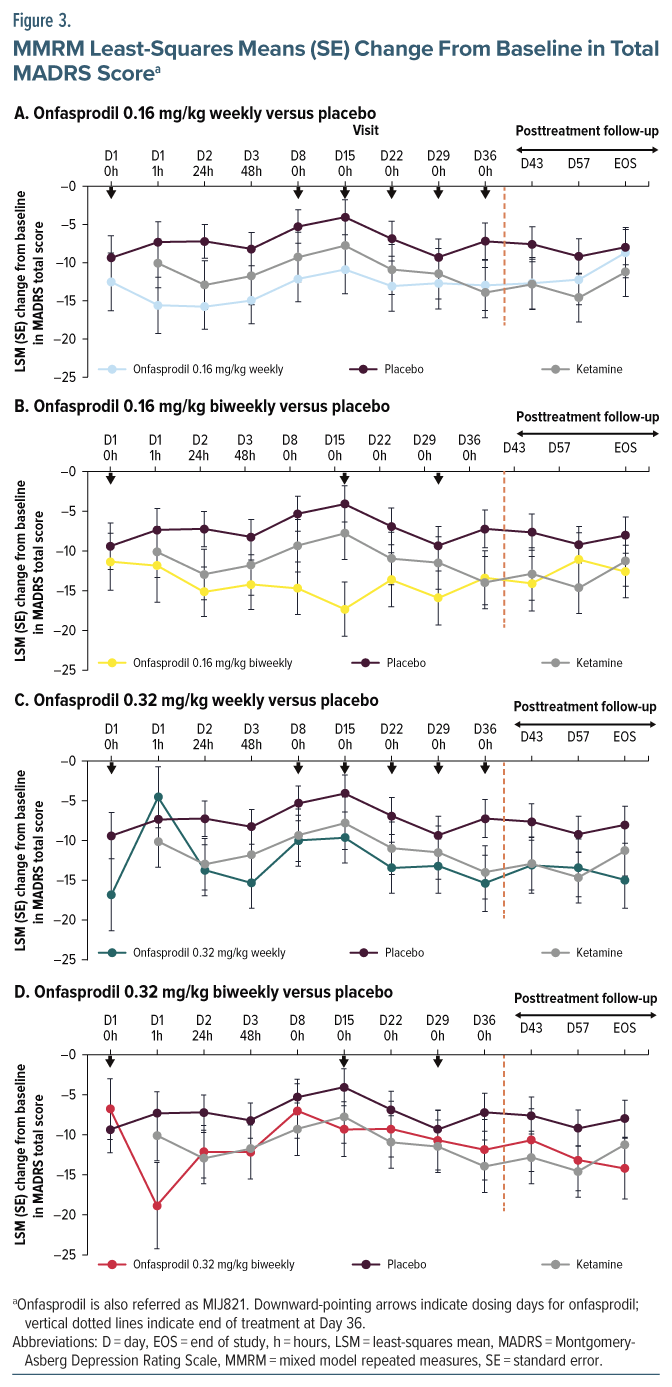
The study enrolled adults (aged 18–65 years) with MDD and prior failure of ≥2 standard antidepressants (where 2 of the failed treatments were different antidepressants, at least 1 of which was being taken in the current depressive episode) of adequate dose and ≥8 weeks duration in a major depressive episode (per DSM-5 criteria) and a Montgomery-Asberg Depression Rating Scale (MADRS) score ≥24.10,11 Details of exclusion criteria are provided in Supplementary Appendix 1.
The study was conducted in accordance with ICH E6 Guideline for Good Clinical Practice as per the Declaration of Helsinki. Study protocol was reviewed and approved by the Independent Ethics Committee or Institutional Review Board for each center. All participants provided written informed consent before study initiation.
Study End Points and Assessments
The primary outcome was change from baseline in the MADRS total score at 24 hours after single-dose administration. The MADRS is a clinician-rated scale designed to measure depression severity and detect changes due to antidepressant treatment. The primary end point was analyzed using an analysis of covariance (ANCOVA) model with treatment as a group factor and baseline MADRS score as a covariate. The 2-sided 80% confidence intervals (CIs) and 1-sided P values were calculated for the treatment differences (each onfasprodil dose versus placebo). Other treatment comparisons (eg, ketamine versus placebo; ketamine versus onfasprodil pooled dose group) were also estimated from the described ANCOVA model; however, the study was not formally powered for these comparisons.
The key secondary outcomes assessed were change from baseline in MADRS score at 48 hours after the first dose and at 6 weeks after repeated dose administration. In addition, the change from baseline in the total MADRS score was analyzed using the mixed-effects models for repeated measures (MMRM). The MMRM model included the fixed, categorical effects of treatment, time (at all planned time points, including but not limited to 24 hours, 48 hours after the first dose, and after the last dose [Day 36]), and treatment × time interaction, as well as the continuous, fixed covariates of baseline score, and baseline score × time interaction. Other secondary efficacy end points were analyzed using the MMRM approach, in the same way as the MADRS total score as described above.
Of particular interest were the dissociative effects, which were measured using the Clinical-Administered Dissociative States Scale (CADSS) questionnaire and the Dissociative Experiences Scale (DES) throughout the study.12,13 Pharmacokinetics, safety, and tolerability were also assessed. Please see Supplementary Appendix 2 for detailed methodology and results of other secondary end points.
All the participants who received at least 1 dose of the study drug were evaluated for safety and adverse events (AEs) including dissociative AEs of interest such as dissociation (environmental perception disturbance or foggy thoughts), amnesia (memory loss), or AEs such as sedation and vomiting.
Please refer to Supplementary Appendix 3 for details of concomitant and prohibited medication.
Statistical Analysis and Sample Size Calculation
Data were analyzed using the analysis sets (see Supplementary Appendix 4). A sample size of 66 patients was planned to be randomized among 6 treatment groups in a 18:12:9:9:9:9 ratio (n = 18 placebo; n = 12 ketamine; n = 9 onfasprodil 0.16 mg/kg weekly; n = 9 onfasprodil 0.16 mg/kg biweekly; n = 9 onfasprodil 0.32 mg/kg weekly; n = 9 onfasprodil 0.32 mg/kg biweekly), which was considered sufficient to achieve the trial objectives. The study investigated 2 primary comparisons at 24 hours after single-dose administration: onfasprodil 0.16 mg/kg vs placebo and onfasprodil 0.32 mg/kg vs placebo. Data from patients assigned to the same dose, but a different regimen were pooled for the purpose of treatment comparison. Based on prior data,1 the standard deviation (SD) of change from baseline to 24 hours after start of first infusion in the total MADRS score was estimated to be 10 points. Assuming the true mean difference (onfasprodil minus placebo) as 8 points (standardized treatment effect size = 0.8), data from 18 evaluable patients per group would provide ∼86% power to detect statistically significant treatment differences using 1-sided α = .10.
RESULTS
Patient Disposition
Overall, 72 patients were randomized, with 2 patients discontinuing from the study before receiving study treatment (Figure 1B). Of the remaining 70 participants, 53 (75.7%) completed the study. The most common reason for discontinuing treatment was subject decision (11 [15.7%]). The mean (SD) age of patients treated in the study was 47.7 (11.3) years. A majority of the participants were White/European American (56%) or Black/African American (41%) (Table 1).
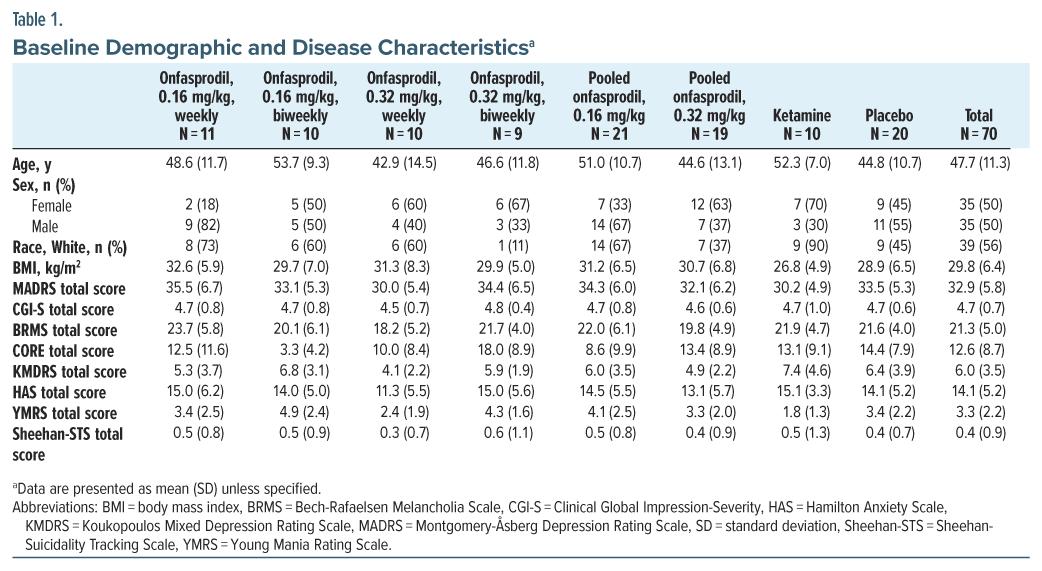
Primary End Point
The study met the primary outcome, a change from baseline in the MADRS total score at 24 hours after single-dose administration. Statistically significant and clinically relevant differences were observed between each of the onfasprodil groups (0.16 and 0.32 mg/kg) and placebo at 24 hours after start of infusion, and the efficacy was maintained in both pooled groups through 48 hours (Figure 2). The adjusted arithmetic mean difference between the onfasprodil 0.16 mg/kg group and the placebo group was −8.25 (P = .0013). The adjusted arithmetic mean difference between the onfasprodil 0.32 mg/kg group and the placebo group was −5.71 (P = .0196). At 24 hours after start of infusion, the ketamine treatment group had a statistically significant lower total MADRS score than the placebo treatment group (adjusted mean difference: −5.67, P = .0461) (Supplementary Table 1). At 48 hours after start of first infusion, a statistically significant decrease in the total MADRS score for both pooled onfasprodil groups and the ketamine group versus placebo was maintained. The magnitude of mean decreases in the total MADRS score in the onfasprodil groups (pooled onfasprodil 0.16 mg/kg: −14.94; pooled onfasprodil 0.32 mg/kg: −15.25) was less pronounced than that observed in the ketamine group (−18.89), but the difference between the onfasprodil groups and ketamine was not statistically significant (Figure 2; Supplementary Table 2).
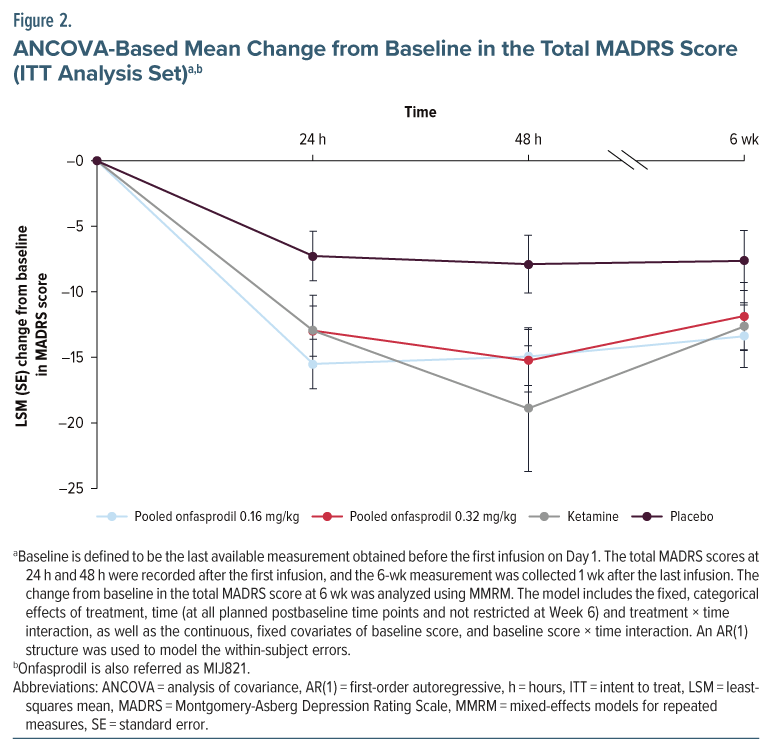
Secondary Efficacy Results
At Week 6 (Day 43 in Figure 3; see also Supplementary Table 3), there was a statistically significant (P < .10) decrease in the total MADRS score for 2 of the 4 onfasprodil treatment groups: onfasprodil 0.16 mg/kg biweekly group (Figure 3B) and onfasprodil 0.32 mg/kg weekly group (Figure 3C) versus the placebo group. On MMRM analysis, the adjusted arithmetic mean difference (80% CI; P value) between the onfasprodil 0.32 mg/kg weekly treatment group and the placebo treatment group was −5.42 (−10.83 to −0.02; P=.0993), and between the onfasprodil 0.16 mg/kg biweekly treatment group and placebo, it was −6.46 (−11.78 to −1.15; P = .0598) (Supplementary Table 4). Thus, the onfasprodil 0.16 mg/kg biweekly group demonstrated the greatest overall MADRS benefit compared with placebo.
The pharmacokinetic methods and results are presented in Supplementary Appendix 5. The Supplementary Figure 1 showed plasma onfasprodil concentrations over time after the first infusion.
Safety
The CADSS total score in the placebo treatment group was low and remained stable throughout the study (Supplementary Table 5). The CADSS total score for the onfasprodil 0.16 mg/kg weekly and biweekly treatment groups was greater than baseline values from the end of the first infusion up to 24 hours after start of infusion, and up to 48 hours after start of infusion for the onfasprodil 0.32 mg/kg weekly and biweekly treatment groups. In the ketamine treatment group, the CADSS total score reached the peak value at 1 hour after the start of infusion (mean change from baseline: 10.30) and then returned to baseline level at 24 hours after the start of infusion.
The DES total score was relatively low in each treatment group and was stable throughout the study, except for higher values in the onfasprodil 0.32 mg/kg biweekly treatment group from baseline to 24 hours after the start of infusion.
At 24 hours, 48 hours, and Week 6 after the start of first infusion, the median score for both the Sheehan-Suicidality Tracking Scale (STS) suicidal behavior subscale and the Sheehan-STS suicidal ideation subscale was 0 in each treatment group.
The incidence of both overall AEs and treatment-emergent AEs (TEAEs) among the onfasprodil-treated groups and the ketamine treatment group was higher than in the placebo treatment group. The proportion of patients who reported at least 1 AE in the pooled onfasprodil 0.16 mg/kg and pooled onfasprodil 0.32 mg/kg was 61.9% (13/21) and 68.4% (13/19), compared to 60.0% (6/10) in the ketamine and 35.0% (7/20) in the placebo group, respectively (Table 2). Most of the AEs were mild in intensity. Moderate and severe AEs were reported in 18.6% and 14.3% of patients, respectively (see Supplementary Table 6; see Supplementary Tables 7–9 for more details on the incidence of AEs by system organ class, preferred terms, and incidence of AEs of interest). One patient in the onfasprodil 0.32 mg/kg biweekly treatment group had a life-threatening AE (suicidal threat), which was not considered to be related to the study treatment.
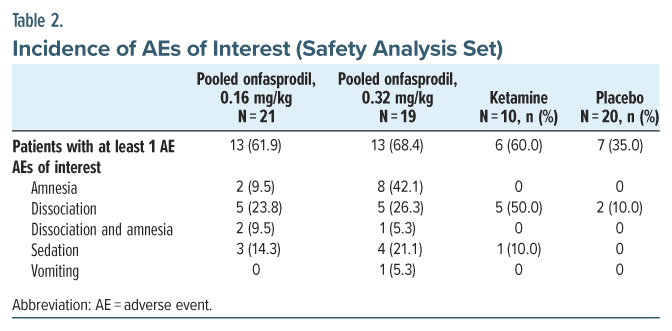
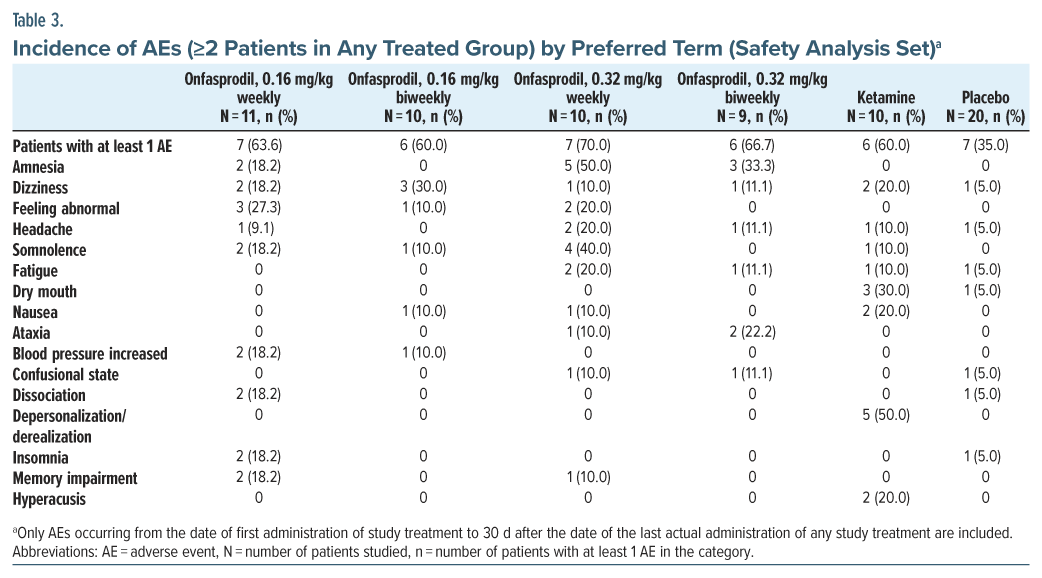
Treatment-emergent AEs were reported in 45.5% (5/11) of patients in the onfasprodil 0.16 mg/kg weekly group, 50.0% (5/10) of patients in the onfasprodil 0.16 mg/kg biweekly group, 70.0% (7/10) of patients in the onfasprodil 0.32 mg/kg weekly group, and 55.6% (5/9) of patients in the onfasprodil 0.32 mg/kg biweekly group, compared to 60.0% (6/10) of patients in the ketamine group and 25.0% (5/20) of patients in the placebo group. The most common TEAEs (>10%) reported among the onfasprodil-treated groups included dizziness, amnesia, somnolence, and feeling abnormal (Table 3). The most common side effects with ketamine were depersonalization/derealization, dry mouth, and dizziness.
The maximum time to onset of significant AEs was 4.4 hours after the start of infusion in patients treated with onfasprodil (sedation) (Supplementary Table 10). All the TEAEs of interest were resolved within a few hours after onset. The maximum time to resolution after the onset of AE was 9.2 hours in patients treated with onfasprodil (Supplementary Table 11). Short periods of amnesia were reported in 10 patients treated with onfasprodil; the time to onset ranged from 0 to 0.7 hours after start of infusion, and the time to resolution ranged from 0.7 to 9.2 hours after onset. Dissociation was reported in 10 patients treated with onfasprodil (5 [23.8%] in the pooled onfasprodil 0.16 mg/kg group and 5 [26.3%] in the pooled onfasprodil 0.32 mg/kg group); the time to onset ranged from 0 to 0.7 hours after the start of infusion, and the time to resolution ranged from 1.6 to 7.0 hours after onset. In the ketamine group, dissociation was reported in 5 (50.0%) patients, the time to onset ranged from 0.1 to 0.2 hours after the start of infusion, and the time to resolution ranged from 0.8 to 2.3 hours after onset.
Serious AEs, including asthma, atrial fibrillation, depression, suicide threat, and suicide attempt, were reported in 5 patients (7.1%): 1 in the onfasprodil 0.16 mg/kg biweekly group, 3 in the onfasprodil 0.32 mg/kg biweekly group, and 1 in the placebo group. None were considered related to the study treatment. Four patients discontinued the study treatment due to AEs, 1 in the onfasprodil 0.16 mg/kg weekly group (blood potassium decreased, alanine aminotransferase [ALT] increased, aspartate aminotransferase [AST] increased, and γ-glutamyl transferase increased), 2 in the onfasprodil 0.32 mg/kg biweekly group (atrial fibrillation and major depression), and 1 in the placebo group (suicidal ideation). No deaths were reported in the study.
DISCUSSION
This study is the first randomized clinical trial that assesses the potential efficacy of a novel NR2B selective negative allosteric modulator, onfasprodil (MIJ821), versus placebo, showing benefit at 24 hours and 48 hours, and persisting up to 6 weeks at lower doses. In a small subgroup, it also compared this agent to the standard NMDA receptor blocker, ketamine, with similar apparent efficacy results. This comparison provides initial data on relative efficacy and safety of this novel agent compared with the most commonly used NMDA receptor inhibitors currently available for clinical care. This proof-of-concept trial demonstrates onfasprodil may be effective for the rapid reduction of depressive symptoms in patients with TRD, with mild dissociative side effects that resolve rapidly. Onfasprodil showed a significantly improved MADRS total score at 24 hours after start of infusion compared with placebo, and efficacy was maintained at 48 hours. The largest and the most consistent benefit versus placebo was observed with the lower dose onfasprodil 0.16 mg/kg biweekly treatment regimen, the effects of which lasted at least 2 weeks.
In this study, the difference between onfasprodil-treated groups and placebo was 5 points or more, which is considered a clinically relevant difference in change in depression.14,15 Onfasprodil demonstrated greater improvements than placebo (0.16 mg/kg pooled group: 8.25-point greater reduction versus placebo) in the MADRS total score at 24 hours. While comparisons across different trials should be done with caution, due to different samples and different methods, the effect size difference between onfasprodil and placebo in this study was consistent with a prior study with a drug with a similar mechanism, traxoprodil (CP101,606) (8.4-point greater reduction versus placebo),6 and prior phase 2 trials of high dose of esketamine (esketamine 84 mg [both periods combined]: 9.0-point greater reduction versus placebo).16
These effect sizes differ from the smaller drug versus placebo effect sizes (2–4 points on MADRS) seen in most studies of selective serotonin reuptake inhibitors17 and phase 3 esketamine trials.18 The rapid onset of action and continued numerical benefit in total MADRS score at Week 6 suggests that onfasprodil may bridge the efficacy gap created by the delayed onset of action of standard antidepressants.
This study employed a less stringent 1-tailed P value than the typical 2-tailed P value that is more commonly seen in registration trials. The reason for this is that this was a proof-of-concept trial for the purpose of the determining if further development is warranted and at what dose and frequency. This is why 2 doses (0.16 and 0.32 mg/kg) at 2 frequencies each (weekly and biweekly) were employed. Using a 1-tailed P value is not unusual in early-stage clinical trials given that sample sizes are usually low and only 1 outcome is meaningful.2 In this case, the only meaningful outcome is if 1 or more doses and frequencies separate from placebo. As noted by Dahlberg et al, “(a) threshold of 0.05 is thought to be the conventional type I error rate; but in fact, the origin of this threshold is arbitrary, and in practice designs often have lower or higher false-positive thresholds depending on design features such as…phase of development.”3 Two-sided P values will be used in future registration trials should they occur.
In the present study, although most patients had low scores in the MADRS suicidal category at baseline, a reduction in suicidal ideation item of the MADRS was seen immediately after the first dose of each study treatment, which remained steady throughout the study with no increased risk. Therefore, onfasprodil may have the potential to improve suicidal symptoms, and further studies in MDD patients with serious suicidal ideation are needed to determine short- and long-term benefits, an effect that is consistent with literature reports for ketamine and esketamine.19–21
Similar to other NR2B-selective antagonists,22 the pharmacodynamic effects of onfasprodil appear to be sustained beyond the level and duration of drug exposure indicated by the pharmacokinetic parameters. While the half-life of onfasprodil is approximately 7 hours, clinical efficacy was maintained for up to 2 weeks. Interestingly, previous studies have shown ketamine and Ro 25-6981 (another NMDA NR2B NAM like onfasprodil) can demonstrate effects on protein synthesis and synaptogenesis.23
Ketamine is a nonselective NMDA receptor blocker that can produce significant adverse effects including dissociative and psychomimetic effects. The data with onfasprodil indicate a favorable safety and tolerability profile with few patients discontinuing due to TEAEs. This tolerability may be attributable to the NMDA receptor subtype selectivity and mechanism of action of this class of molecules.24,25
The most frequently reported treatment-related AEs with onfasprodil were dizziness, amnesia, and somnolence. Dissociative-type side effects were mild with onfasprodil. The maximum increase in CADSS total score was higher in the ketamine group (mean maximum change from baseline was 10.30) than in the onfasprodil treatment groups (mean maximum change from baseline: ≤5).
Of note, all the treatment-related AEs of interest resolved within a few hours after onset, and there were no clinically relevant changes in clinical laboratories, vital signs, or ECGs. Overall, onfasprodil was well tolerated across all dosing regimens.
Several limitations of the study should be considered in the interpretation of the findings. The sample size was relatively small, and the trial was conducted in a limited number of research sites. The dropout rate in the acute treatment phase ranged from 10% in the ketamine group to 33.3% in the onfasprodil 0.32 mg/kg weekly groups. Ketamine was administered only once per week and how this compares to the more standard twice-weekly initial dosing is unknown. Ketamine was also administered at a fixed dose rather than the more customary 0.5 mg per kg over 40 minutes. Therefore, the plasma concentrations might have been lower for some patients with higher body mass indexes. Nonetheless, there was a statistically significant and clinically meaningful difference between ketamine and placebo. A greater difference might have been achieved with more conventional dosing. However, a significant difference between ketamine and placebo assured sufficient assay sensitivity, ensuring that the study was internally valid.
Despite the small sample size, this proof-of-concept study suggests that selected dosing regimens of onfasprodil may be effective and well-tolerated in patients with TRD, with rapid onset of action (24-hours) with evidence of antidepressant effects to be maintained at Week 6, particularly for the lower-dose group. Further studies with larger sample sizes are necessary to confirm these preliminary findings.
Article Information
Published Online: August 6, 2025. https://doi.org/10.4088/JCP.23m15246
© 2025 Physicians Postgraduate Press, Inc.
Submitted: December 31, 2023; accepted February 28, 2025.
To Cite: Shelton RC, Litman RE, Hassman H, et al. Rapid onset and sustained efficacy of onfasprodil (MIJ821), a novel NR2B negative allosteric modulator, in patients with treatment-resistant depression: a phase 2, randomized, placebo-controlled, proof-of concept study. J Clin Psychiatry 2025;86(3):23m15246.
Author Affiliations: Department of Psychiatry and Behavioral Neurobiology, Heersink School of Medicine, University of Alabama at Birmingham, Birmingham, Alabama (Shelton); Georgetown University Medical School, Washington, DC (Litman); Hassman Research Institute, Marlton, New Jersey (Hassman); Collaborative Neuroscience Network Garden Grove, Garden Grove, California (Walling); Instituto Internacional de Neurociencias Aplicadas Barcelona, Barcelona, Spain (Montalbán); Hospital Universitari Son Espases, Servicio de Psiquiatría, UIB, IdISBa, Palma de Mallorca, Spain (Coll); Psychiatric Medicine Associates, LLC, Skokie, Illinois (Zajecka); Novartis Pharmaceuticals Corporation, East Hanover, New Jersey (Sverdlov); Novartis Biomedical Research, Cambridge, Massachusetts (Gomez-Mancilla, Healy, Shanker, Berkheimer, Cha, Ghaemi); Novartis Pharma AG, Basel, Switzerland (Gomez-Mancilla, Faller, Serban); McGill University, Montreal, Québec, Canada (Gomez-Mancilla); Beam Therapeutics, Cambridge, Massachusetts (Shanker); Novartis Pharma S.A.S., Rueil-Malmaison, France (von Raison); Tufts University School of Medicine, Boston, Massachusetts (Ghaemi); Harvard Medical School, Boston, Massachusetts (Ghaemi).
Corresponding Author: Mark P. Healy, PhD, Global Discovery Chemistry, Novartis Biomedical Research, 22 Windsor St, Cambridge, MA 02478 ([email protected]).
Relevant Financial Relationships: Dr Shelton received grants from the National Institutes of Health, Patient-Centered Outcomes Research Institute, AbbVie Inc, Alkermes, Inc, Allergan, Plc, Alto Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb Company, Denovo Biopharma, Equulus Therapeutics, InMune Bio, Intra-cellular Therapies, Johnson & Johnson, LivaNova PLC, Navitor Pharmaceuticals, Neurocrine Biosciences, Novartis Pharmaceuticals, Otsuka Pharmacetical Company, Sunovion Pharmaceuticals, Syndeio Biosciences, Takeda Pharmaceuticals; has been a paid consultant for Abbvie Acadia Pharmaceuticals, Allergan, Plc, Boehringer Ingelheim, Cerecor, Inc, Denovo Biopharma, Johnson & Johnson, Neurorx, Novartis AG, Otsuka Pharmaceuticals, Seelos Therapeutics, Inc, Sunovion Pharmaceuticals, and Takeda Pharmaceuticals; and receives royalties from Springer Nature and Wolters Kluwer N.V. Dr Litman received grants from Allergan, BioXcel, Concert, Janssen, Karuna, Otsuka, Roche, Sage, Shenox, Sunovion, Takeda, Teva, and Vistagen, speaks for AbbVie and Allergan, and serves on the scientific advisory board for Terran BioSciences. He is a Clinical Associate Professor at the Georgetown University Medical School. Dr Walling reports grants from AbbVie, Acadia, Alkermes, Allergan, Avanir, Biogen Boehringer Ingelheim, Cerevel, CoMentis, Intra-Cellular Therapies, Indivior, Janssen, Johnson & Johnson PRD, Lundbeck, Lupin, Novartis, Noven, Otsuka, Prothena, Pfizer, Roche, Sunovion, and Takeda and personal fees from Janssen, Otsuka, Boehringer Ingelheim, Biogen, and Lyndra. Dr Ros Montalbán has received fees as speaker from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Esteve, Exeltis, Janssen-Cilag, Juste, Lilly, Lundbeck, Organon, Otsuka, Pfizer, Rovi, Servier; has received financial compensation for his participation as a board member of Bristol Myers Squibb, Janssen-Cilag, Lundbeck. Organon, Otsuka, Pfizer; and has received research funding from AstraZeneca, Janssen-Cilag. Lundbeck, Novartis, Otsuka, and Roche. Dr Salvà Coll received honoraria from Novartis Inc, Janssen Pharmaceutica, Roche, AstraZeneca, and Otsuka Pharmaceuticals. Dr Zajecka received grants from Alkermes, Allergan, Boehringer-Ingelheim, Cheryl T. Herman Foundation, Elminda Ltd, Hoffman-LaRoche, Intracellular Therapies, Janssen/ Johnson & Johnson, LivaNova, Lundbeck, Novartis, Otsuka, Praxis, Sage therapeutics, Takeda, COMPASS, and Neurocrine; received advisory board fees from Alkermes, Alphasigma USA, Inc, Elminda, Ltd, Janssen/Johnson & Johnson, LivaNova, Lundbeck, Seelos Therapies, and Takeda; and received other financial support from Cheryl T. Herman Foundation. Dr Gomez-Mancilla was an employee of Novartis Pharma AG at the time of this study and is a current employee of STALICLA. Dr Shanker was an employee of Novartis at the time of this study and is a current employee of Beam Therapeutics. Dr Ghaemi was an employee of Novartis Biomedical Research at the time of this study and is currently Professor of Psychiatry at Tufts University and Lecturer on Psychiatry at Harvard Medical School, Boston, Massachusetts. Drs Sverdlov, Berkheimer, Faller, Healy, von Raison, and Serban are employees and own stock in Novartis. Dr Cha was an employee of Novartis Biomedical Research at the time of this study and is a current employee of Latus Bio. Dr Hassman reports no financial relationships with commercial interests.
Funding/Support: This study was funded by Novartis Biomedical Research, Cambridge, MA.
Role of the Sponsor: The study was only sponsored and supported by Novartis Biomedical Research, Cambridge, Massachusetts.
Previous Presentation: Most of the data in the article have not been presented previously. Aspects (efficacy and safety) were presented at American Psychiatric Association (Virtual); May 1–3, 2021, and 29th European Congress of Psychiatry (Virtual); April 10–13, 2021.
Acknowledgments: The authors thank the patients and center participants and others involved in the study. The authors also thank all former employees or consultants with Mnemosyne Pharmaceuticals Inc/Luc Therapeutics/Cadent Therapeutics, for their contributions to the discovery of onfasprodil and Rohita Sri Gattoju, MSc, and Karanam Ananda Krishna, PhD, from Novartis Healthcare Pvt Ltd, Hyderabad, India, for providing medical writing support for the manuscript, which was funded by Novartis Pharma AG, Basel, Switzerland, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Supplementary Material: Available at Psychiatrist.com.
Clinical Points
With the limitation of small sample sizes per subgroup arm, this proof-of-concept study demonstrates the following:
- Rapid onset of action of onfasprodil and sustained benefit with repeated dosing vs placebo, and efficacy similar to ketamine in a small subgroup, suggesting possible therapeutic utility in the treatment-resistant depression population.
- An overall good safety profile that was well tolerated.
- Greater efficacy at the lowest dose at the lowest frequency of onfasprodil vs other arms.
References (25)

- Gaynes BN, Lux L, Gartlehner G, et al. Defining treatment-resistant depression. Depress Anxiety. 2020;37(2):134–145. PubMed CrossRef
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. PubMed CrossRef
- Katalinic N, Lai R, Somogyi A, et al. Ketamine as a new treatment for depression: a review of its efficacy and adverse effects. Aust N Z J Psychiatry. 2013;47(8):710–727. PubMed CrossRef
- Krystal JH, Kavalali ET, Monteggia LM. Ketamine and rapid antidepressant action: new treatments and novel synaptic signaling mechanisms. Neuropsychopharmacology. 2024;49(1):41–50. PubMed CrossRef
- Molero P, Ramos-Quiroga JA, Martin-Santos R, et al. Antidepressant efficacy and tolerability of Ketamine and Esketamine: a critical review. CNS Drugs. 2018;32(5):411–420. PubMed CrossRef
- Preskorn SH, Baker B, Kolluri S, et al. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28(6):631–637. PubMed CrossRef
- Kawai M, Ando K, Matsumoto Y, et al. Discovery of (-)-6-[2-[4-(3-fluorophenyl)-4-hydroxy-1-piperidinyl]-1-hydroxyethyl]-3,4-dihydro-2(1H)-quinolinone-a potent NR2B-selective N-methyl D-aspartate (NMDA) antagonist for the treatment of pain. Bioorg Med Chem Lett. 2007;17(20):5558–5562. PubMed CrossRef
- Löscher W, Rogawski MA. In: Lodge D, Danysz W, Parsons GC, et al, eds. Chapter 3: Epilepsy. From: Ionotropic Glutamate Receptors as Therapeutic Targets. FP Graham Publishing Co;2002:1–42.
- Gomez-Mancilla B, Levy JA, Ganesan S, et al. MIJ821 (onfasprodil) in healthy volunteers: first-in-human, randomized, placebo-controlled study (single ascending dose and repeated intravenous dose). Clin Transl Sci. 2023;16(11):2236–2252. PubMed CrossRef
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. American Psychiatric Publishing, Inc; 2013.
- Khan A, Khan SR, Shankles EB, et al. Relative sensitivity of the montgomery-asberg depression rating scale, the Hamilton depression rating scale and the clinical global impressions rating scale in antidepressant clinical trials. Int Clin Psychopharmacol. 2002;17(6):281–285. PubMed CrossRef
- Bernstein EM, Putnam FW. Development, reliability, and validity of a dissociation scale. J Nerv Ment Dis. 1986;174(12):727–735. PubMed CrossRef
- Bremner JD, Krystal JH, Putnam FW, et al. Measurement of dissociative states with the Clinician-Administered Dissociative States Scale (CADSS). J Trauma Stress. 1998;11(1):125–136. PubMed CrossRef
- Montgomery SA, Möller HJ. Is the significant superiority of escitalopram compared with other antidepressants clinically relevant? Int Clin Psychopharmacol. 2009;24(3):111–118. PubMed CrossRef
- Hengartner MP, Jakobsen JC, Sørensen A, et al. Efficacy of new-generation antidepressants assessed with the Montgomery-Asberg Depression Rating Scale, the gold standard clinician rating scale: a meta-analysis of randomised-placebo controlled trials. PLoS One. 2020;15(2):e0229381. PubMed CrossRef
- Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139–148. PubMed CrossRef
- Thase ME, Larsen KG, Kennedy SH. Assessing the “true” effect of active antidepressant therapy v. placebo in major depressive disorder: use of a mixture model. Br J Psychiatry. 2011;199(6):501–507. PubMed CrossRef
- US Food and Drug Administration. Center for Drug Evaluation and Research. Clinical Review: Application Number: 211243Orig1s000. Accessed January 7, 2025. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/211243Orig1s000MedR.pdf
- Osby U, Brandt L, Correia N, et al. Excess mortality in bipolar and unipolar disorder in Sweden. Arch Gen Psychiatry. 2001;58(9):844–850. PubMed CrossRef
- Reinstatler L, Youssef NA. Ketamine as a potential treatment for suicidal ideation: a systematic review of the literature. Drugs R D. 2015;15(1):37–43. PubMed CrossRef
- Canuso CM, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine for the rapid reduction of symptoms of depression and suicidality in patients at imminent risk for suicide: results of a double-blind, randomized, placebo-controlled study. Am J Psychiatry. 2018;175(7):620–630. PubMed CrossRef
- Ibrahim L, Diaz Granados N, Jolkovsky L, et al. A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J Clin Psychopharmacol. 2012;32(4):551–557. PubMed CrossRef
- Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. PubMed CrossRef
- Kemp JAKJ, Gill R. NMDA receptor antagonists and their potential as neuroprotective agents. In: Handbook of Experimental Pharmacology Volume 141. Ionotropic Glutamate Receptors in the CNS; Springer;1999:495–527.
- Gogas KR. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr Opin Pharmacol. 2006;6(1):68–74. PubMed CrossRef
This PDF is free for all visitors!





