
Abstract
Objective: Postpartum depression (PPD) is a debilitating mood disorder with peripartum onset. Current treatment options are limited in PPD. GH001 is a synthetic inhalation formulation of the psychoactive molecule mebufotenin (5-MeO-DMT). This trial investigated the preliminary efficacy and safety of GH001 in adult females with PPD.
Methods: This phase 2a, proof-of-concept, open-label trial enrolled women aged 18–45 years (March 2023 to August 2024) with a Mini-International Neuropsychiatric Interview–confirmed diagnosis of major depressive disorder with peripartum onset. Patients had Montgomery-Asberg Depression Rating Scale (MADRS) scores of ≥28 at baseline. GH001 was administered as an individualized dosing regimen of up to 3 escalating doses (6, 12, and 18 mg) on day 1. The primary end point was the change in MADRS total score from baseline to day 8. Secondary end points included antidepressant response (≥50% reduction), remission (MADRS total score ≤10), and safety and tolerability of GH001.
Results: Ten patients were enrolled. Mean baseline MADRS total score was 36.7 (SD = 4.8). Mean MADRS total score change from baseline to day 8 was −35.4 points (SD = 5.5; P < .0001). All patients achieved response and were in remission on day 8, which was first observed 2 hours after their final dose on day 1. Inhalation of GH001 was well tolerated, and no serious adverse events (AEs) were reported. All treatment-emergent AEs were mild or moderate, with headache as the most frequently reported AE.
Conclusion: GH001 demonstrated rapid and significant improvements in depressive symptoms and remission of PPD with an acceptable safety profile and parallel improvements across secondary end points.
Trial Registration: ClinicalTrials.gov identifier: NCT05804708; EudraCT identifier: 2021-006879-42
J Clin Psychiatry 2026;87(3):25m16284
Author affiliations are listed at the end of this article.
From the Editors

Are you a healthcare provider?
Add your NPI to personalize your JCP experience.
Postpartum (or peripartum) depression (PPD) is a common peripartum complication that can have serious consequences for the well-being of the mother and the long-term development of the child.1,2 Epidemiologic studies estimate the global prevalence rate of PPD to be as high as 20%, with up to 30% of diagnosed patients still experiencing symptoms 2 years after giving birth.3–5 PPD is associated with numerous short- and long-term negative outcomes for the mother, child, and the entire family, especially if treatment is delayed and/or insufficient.6–8 Typical antidepressive treatments (eg, selective serotonin reuptake inhibitors [SSRIs]) have a slow onset of action, low remission rates, and chronic or long-lasting side effects that can lead to discontinuation.9,10 Zuranolone, the only treatment for PPD approved by the US Food and Drug Administration (FDA), demonstrates rapid-acting antidepressant effects over a 14-day treatment course; however, clinical use remains low as compared to serotonergic antidepressants.10–13
Mebufotenin (5-methoxy-N,N-dimethyltryptamine [5-MeO-DMT]) is a potent psychoactive molecule that acts as a nonselective serotonin agonist with higher affinity for the 5-HT1A receptor subtype versus the 5-HT2A subtype and a short half-life in plasma.14,15 Early-phase clinical trials of mebufotenin administered via pulmonary inhalation (GH001) demonstrated acceptable safety and tolerability, with an ultrarapid onset of antidepressive effects in patients with treatment-resistant depression (TRD).16,17 This is the first clinical trial of mebufotenin in adult, female patients with PPD, investigating its potential antidepressant effects, safety, and impact on maternal functioning and behavior.
METHODS
Trial Oversight
The clinical trial screened patients from 3 sites in the United Kingdom and the Netherlands and enrolled patients from 1 site in the United Kingdom between March 2023 and August 2024. The trial was conducted in accordance with the International Council for Harmonisation Good Clinical Practice and ethical principles of the Declaration of Helsinki.18 The regulatory authority and research ethics committee for each trial site approved the protocol before patient enrollment. All patients provided their written informed consent after being fully informed about the purpose of the trial and after procedure(s) and possible side effects were fully explained. The sponsor (GH Research, Dublin, Ireland) designed and funded the trial and supported medical writing assistance for drafting the manuscript under direction from the authors.
Patients
This phase 2a, proof-of-concept, single-arm, open-label trial enrolled female outpatients aged 18–45 years who met the Mini-International Neuropsychiatric Interview19 (v 7.0.2) diagnostic criteria for major depressive disorder with peripartum onset, were >4 weeks postpartum at dosing and ≤12 months postpartum at screening, and had a Montgomery-Asberg Depression Rating Scale (MADRS) total score of ≥28, reflecting moderate-to-severe depressive symptoms. Antidepressants, antipsychotics, psychedelics, dissociatives, and any medication with monoamine oxidase inhibitor activity were prohibited during the trial period and within 2 weeks (or 5 half-lives prior to baseline, whichever was longer). Any decision to discontinue medication was made by patients and investigators, based on clinical judgment, with involvement of patients’ physicians or psychiatrists; no medication was discontinued for the sole purpose of allowing patients to participate in the trial. Any tapering/washout schedule was performed per clinical practice, with support from research site staff. Initiation or modification of psychotherapy during the trial was prohibited. Patients either had to have ceased lactating at screening or, if still lactating or actively breastfeeding, must have agreed to temporarily cease breastfeeding from just prior to dosing through 24 hours after the last dose. Additional details including the full inclusion and exclusion criteria are provided in the Supplementary Appendix.
Trial Visits
The trial consisted of a screening period; a day 1 visit where eligibility was reconfirmed; a day 1 visit where patients received the study drug, GH001; a day 2 follow-up visit; and the end of trial visit on day 8. The trial was conducted under the supervision of qualified health care professionals, providing psychological support per standard of care, but without any planned psychotherapeutic intervention before, during, or after dosing, as per recent FDA recommendations.20 On the same day of dosing (day 1), patients were discharged once all trial assessments were completed and once they were deemed discharge-ready as assessed using a proprietary assessment, the Clinical Assessment of Discharge Readiness (CADR), administered by a health care professional.
Study Drug
GH001, an inhalation formulation of synthetic mebufotenin, was administered using the Volcano Medic 2 Vaporization System (Storz & Bickel, Germany). The system vaporizes the formulation to produce an aerosol, which is collected in a detachable balloon for inhalation by the patient. Prior to GH001 administration, patients were trained in the inhalation technique and were prepared for the potential psychoactive effects of the study drug by the trial physician. Patients received GH001 as an individualized dosing regimen (IDR) of up to 3 escalating doses (6, 12, and 18 mg) on a single day with a 1-hour interval between doses (Figure 1). A second or third dose was administered if the previous dose was well tolerated according to the trial physician’s judgment (based on vital signs and adverse events [AEs]) and if the patient did not achieve an intense psychoactive effect (peak experience [PE], defined as a mean score of ≥75 on the Peak Experience Scale [PES])21 following the previous dose; further details are provided in the Supplementary Appendix.
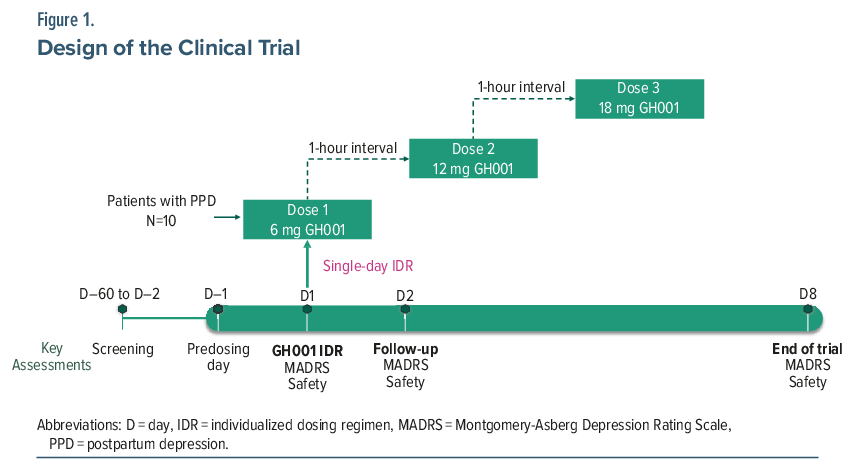
Assessments
The primary end point was mean change in severity of depressive symptoms assessed by MADRS22 total score from baseline to day 8. Secondary efficacy end points included mean change from baseline in MADRS total score at 2 hours after the final dose of the IDR and on day 2; proportion of patients in remission (MADRS total score ≤10); proportion of patients with treatment response (≥50% reduction from baseline MADRS total score); overall disease severity assessed by change from baseline to day 8 on the Clinical Global Impression–Severity (CGI-S) scale23; and maternal functioning and behavior by change from baseline to day 8 in Barkin Index of Maternal Functioning (BIMF)24 total score and functional area scores.
Safety and tolerability were assessed throughout the trial as secondary end points and included the following parameters: incidence of treatment-emergent adverse events (TEAEs) classified according to the Medical Dictionary for Regulatory Activities (MedDRA), version 27.1; vital signs, electrocardiogram, spirometry, standard safety laboratory analyses (hematology, biochemistry, urinalysis), sedation as assessed by the Modified Observer’s Assessment of Alertness and Sedation (MOAA/S),25 psychiatric symptoms as assessed by the Brief Psychiatric Rating Scale (BPRS),26 dissociative symptoms assessed by the Clinician Administered Dissociative States Scale (CADSS),27 suicidality as assessed by the Columbia-Suicide Severity Rating Scale (C-SSRS),28 cognitive effects assessed using the Cambridge Neuropsychological Test Automated Battery (CANTAB),29 and discharge readiness on day 1 assessed using the CADR. Psychoactive effects of the study drug were assessed by the proprietary PES,21 the Challenging Experience Questionnaire (CEQ),30 and the Mystical Experience Questionnaire 30-item version (MEQ30),31 as well as the duration of the psychoactive effects. As an exploratory end point, concentrations of mebufotenin, its psychoactive metabolite bufotenine, and its terminal nonpsychoactive metabolite 5-methoxyindole-3-acetic acid (5-MIAA) were measured in breast milk of lactating patients using liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods. The assigned lower limit of quantification for LC-MS/MS was 0.025 ng/mL. Further details including timing of all assessments can be found in the Supplementary Appendix.
Statistical Methodology
For the primary end point, a P value was calculated using a 1-sample t-test with a 1-sided significance level of α = .025. The trial was originally powered to include 15 patients; with 10 patients, the trial remained adequately powered to detect a clinically meaningful difference. Secondary and exploratory end points were reported using descriptive statistics. The trial was registered on ClinicalTrials.gov (NCT05804708) and EudraCT (2021-006879-42).
RESULTS
Patients
From March 2023 to August 2024, the trial enrolled 10 patients diagnosed with PPD with a mean (SD) age of 31.6 (5.2) years. The mean (SD) duration of their current depressive episode was 30.9 (12.9) weeks, and the mean (SD) parity was 2 (0.9). One patient (10.0%) had received pharmacotherapy for the current depressive episode, and 6 patients (60.0%) had received pharmacotherapy for prior major depressive episodes. The mean (SD) baseline MADRS total score was 36.7 (4.8; n = 10), baseline CGI-S was 4.8 (0.79; n = 10), and baseline BIMF total score was 68.8 (15.6; n = 8) out of a possible total score of 120, indicating a significant degree of functional impairment.24 Additional details of the patient population are outlined in Table 1. All patients completed the trial, with GH001 IDR dosed as follows: 6 mg (n = 1), 6 + 12 mg (n = 7), 6 + 12 + 18 mg (n = 2) (Supplementary Table 3).
Efficacy
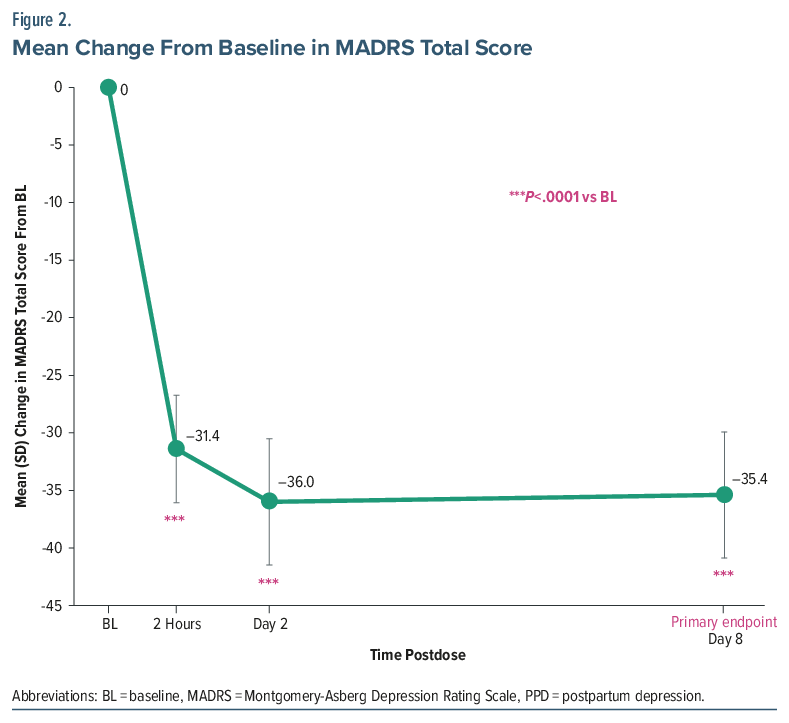
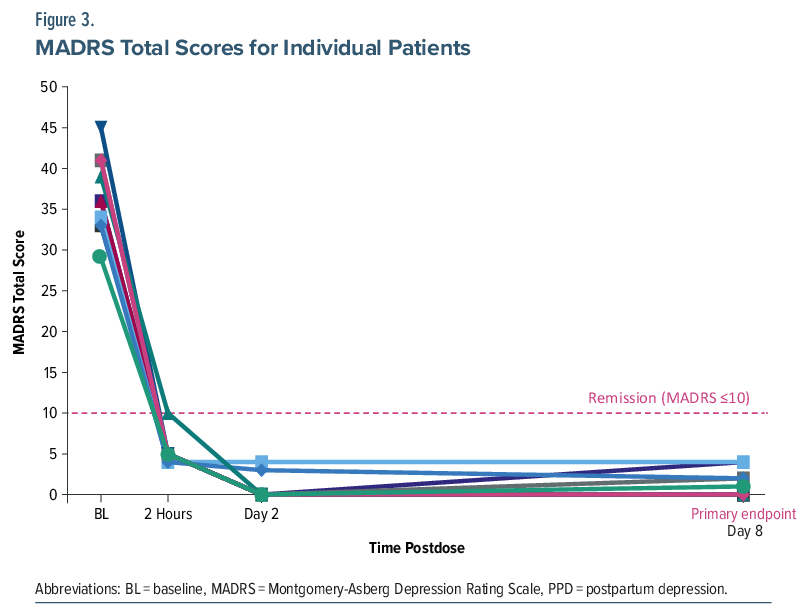
The primary end point was achieved, with a significant mean (95% CI) reduction from baseline to day 8 of −35.4 points (−39.32 to −31.48) in MADRS total score with GH001 treatment (P<.0001), corresponding to an approximate 96% relative reduction from baseline. Significant reductions in MADRS total score were also observed at 2 hours postdose and on day 2 (P <.0001 for both time points; Figure 2). All 10 patients demonstrated large and consistent reductions in MADRS total score at 2 hours postdose, on day 2, and on day 8 (Figure 3). All patients (100%) achieved both response and remission at day 8, as well as at 2 hours postdose and on day 2. All patients had a reduction in the illness severity measured by the CGI-S, with mean (SD) change in score from baseline to 2 hours postdose, day 2, and day 8 of −3.7 (0.82), −3.8 (0.79), and −3.8 (0.83), respectively. At day 8, BIMF total score increased by a mean (SD) of 34.1 (16.10) points (n=8), an approximate 56% improvement. Increases from baseline were observed on day 8 across most BIMF functional areas (Supplementary Table 4).
Psychoactive Effects
Assessed by the PES, 7 of 10 patients (70.0%) achieved a PE. No consistent effects were noted for the CEQ or the MEQ30. The clinician-reported duration of psychoactive effects following GH001 administration showed no clear dose response, with median (range) individual dose durations of 22.0 (5–62), 25.0 (7–37), and 25.0 (10–40) minutes after the 6-, 12-, and 18-mg doses, respectively.
Safety
Thirteen TEAEs were observed in 8 of 10 patients (80.0%), and were mostly mild in severity (87.5%); only 1 patient reported a TEAE with moderate severity (Table 2). Headache was the most reported TEAE (5/10 patients); all other TEAEs occurred in a single patient each. No TEAEs of flashbacks, serious TEAEs, or severe TEAEs were reported, and no patients withdrew from the trial. There was no apparent dose-response relationship with TEAEs; AEs were distributed across dose levels without a clear pattern (Table 2).
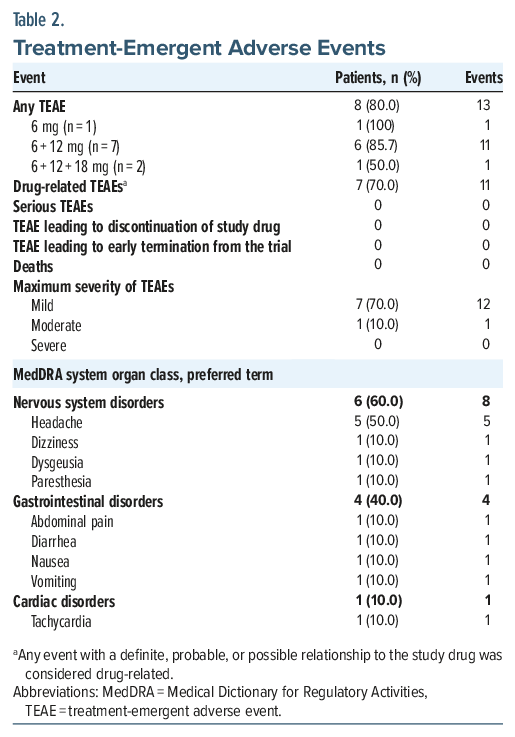
Assessment of psychiatric and psychotic symptoms with the BPRS showed a mean (SD) reduction in BPRS total score from baseline to day 8 of −23.7 (8.34), bringing the day 8 score of 18.1 (0.33) close to the 18-point minimal score of the BPRS. There were no notable or consistent changes from baseline values in clinical laboratory assessments of hematology, chemistry, or urinalysis, and no individual patient had abnormal clinically significant (CS) shifts in any laboratory measurement. Similarly, assessments of vital signs showed no CS changes, and there were no clearly identifiable trends in respiratory rate, SpO2, body temperature, spirometry, or body weight. As in other trials with psychedelics,32–35 transient increases in blood pressure and heart rate were observed after administration of GH001, spontaneously returning to baseline within 20–60 minutes after dosing. Assessed after the psychoactive effects had subsided, there was no worsening of clinician-rated assessments (CADSS and MOAA/S) and no consistent evidence of a GH001-mediated impairment in performance on CANTAB tasks. The proportion of patients reporting suicidal ideation per the C-SSRS was reduced from 3 patients (30.0%) at baseline to none (0%) at discharge (day 1), day 2, and day 8; no patients reported suicidal behavior or nonsuicidal self-injurious behavior at any of the scheduled C-SSRS assessments. Using the CADR, all patients were deemed ready for discharge within the dosing day.
Exploratory Breast Milk Analysis
Of the lactating patients (n = 4), 3 received GH001 6+12 mg and 1 received GH001 6 + 12 + 18 mg as part of the IDR. Mebufotenin levels in breast milk ranged from 0.24 to 3.11 ng/mL at 1 hour postdose, declining to below the limit of quantification (BLQ, 0.025 ng/mL) to 0.04 ng/mL ∼8 hours postdose; all levels were BLQ on days 2 and 8. Bufotenin was BLQ in breast milk at all time points. 5-MIAA levels ranged from 13.9 to 28.5 ng/mL at 1 hour, declining to BLQ to 0.90 ng/mL at ∼8 hours and BLQ on day 8.
DISCUSSION
In this phase 2a, proof-of-concept, open-label trial evaluating the antidepressant effects and safety of GH001 in a group of patients with moderate and severe PPD (mean baseline MADRS >34), the primary end point was met. A significant mean reduction of 35.4 points in MADRS total score (corresponding to an approximate 96% reduction in symptom severity) was found from baseline to day 8, with clinically meaningful effects observed as early as 2 hours postdose and all patients achieving remission, which was maintained through the final assessment on day 8. While cross-study comparisons must be interpreted cautiously, these findings indicate a time to response that is considerably faster than what has been reported for the approved treatment zuranolone and the previously approved but now withdrawn brexanolone.11–13,36,37 The MADRS reduction observed here (−35.4 points) compares favorably with that reported in the brexanolone proof-of-concept randomized controlled trial in PPD (−28.0 points at 60 hours; n = 10).36 Antidepressant efficacy has also been observed with GH001 in TRD, where a placebo-adjusted MADRS reduction of −15.5 points (Cohen’s d = 2.0) was demonstrated in a phase 2b trial,38 and in bipolar II disorder with a current major depressive episode, where a MADRS reduction of −16.8 points was observed in a phase 2a open-label trial.39
Further strengthening the validity of the observed antidepressant effects, parallel improvements were seen in the CGI-S and BPRS scales, with broad improvement across the assessed psychopathological symptoms of the BPRS. On the BIMF scale, GH001 was associated with a 56% improvement on the overall score, with improvements across multiple domains of self-reported maternal functioning, predominantly psychological well-being. GH001 administered via inhalation further demonstrated a favorable safety profile without sedative effects and with same-day discharge from the clinic. The inhalation was well tolerated, and no serious TEAEs were reported. Preliminary results of the pharmacokinetic analysis of breast milk in this trial support a treatment strategy with only a brief interruption of breastfeeding around GH001 dosing. Mebufotenin and its metabolite 5-MIAA were transiently present in breast milk and were both rapidly eliminated by ∼10 hours postdose. Bufotenin was not detected in any breast milk sample.
The pharmacologic profile of GH001 differs from that of currently available treatments for PPD. SSRIs, the established first-line treatment, have an onset of action of 4–6 weeks with side effects including headache, nausea, somnolence, and sexual dysfunction which can impact adherence.10 Zuranolone, an orally formulated γ-aminobutyric acid-A receptor modulator, requires a 14-day dosing schedule with onset over several days.11–13 Common side effects include somnolence, dizziness, and fatigue, with driving restrictions for 12 hours after each dose.40 This trial applied diligent assessment of such effects using specific scales for sedation and posttreatment drowsiness (MOAA/S and CADR) and dissociation (CADSS) and found no signs of adverse effects after the psychoactive effects had subsided, with all patients being discharge-ready shortly after receiving the last dose of study drug and no negative effects on cognitive function. Psychotherapeutic interventions are commonly employed in trials for psychoactive molecules and may increase expectancy and performance biases.20 Psychotherapeutic interventions were not included in the trial design and thus did not contribute to observed treatment benefits with GH001. When designing the trial, there was a focus on patient safety, securing a patient population without significant psychiatric comorbidity, not presently receiving antidepressant treatment, and with the availability of a person to act as a caregiver for the child while the patient took part in the trial. These strict requirements limited the recruitment of patients, so while the trial originally planned to include 15 patients, enrolment was stopped early due to recruitment challenges.
Certain limitations of this trial must be acknowledged including the enrolment of a small number of patients (N = 10), its relatively short follow-up of 1 week, the lack of a control arm, and no blinding. These factors may limit the ability to attribute changes in depressive symptoms exclusively to the investigational treatment. The predominantly White (90%) population, and the fact that 9/10 patients were not receiving pharmacotherapy for their current depressive episode, may further limit the generalizability of these findings to broader, more diverse PPD populations including those on concurrent antidepressant treatment. Additionally, while the magnitude and rapidity of symptom reduction with GH001 are noteworthy, comparisons with other medications for PPD must be made cautiously due to differences in trial populations, methodologies (ie, placebo-controlled trial vs open-label trial), and outcome measures. Mechanistically, mebufotenin is pharmacologically distinct from SSRIs and zuranolone, acting as a direct, nonselective serotonin receptor agonist with preferential 5-HT1A affinity that engages glutamatergic signaling and rapid neuroplasticity, potentially accounting for the rapid onset of antidepressant effects.41 The ultra-short duration of psychoactive effects enables the single-day IDR to be administered during a single supervised visit with same-day discharge, which may support administration in an outpatient setting.
The American Psychiatric Association recommends initiating or continuing pharmacotherapy for patients with PPD, especially those with severe illness, using the minimally effective dose and considering breastfeeding safety.42 Should these findings be replicated in larger, placebo-controlled trials, GH001’s rapid onset, short half-life, and favorable safety profile would fit well into the existing PPD management recommendations. Emerging data from a 6-month open-label extension of a phase 2b trial in TRD suggest that intermittent retreatment with GH001, rather than continuous maintenance pharmacotherapy, may be sufficient to sustain remission; whether a similar approach is applicable in PPD will require investigation in future trials. Given the preliminary improvements in maternal functioning, future research should evaluate GH001’s impact on the mother-infant relationship.
In conclusion, GH001 administered as a single-day IDR in adult female patients with PPD was well tolerated with an acceptable safety profile, with large improvements in depressive symptoms and maternal functioning up to 7 days posttreatment. These results support further investigations of the longer-term efficacy and safety of GH001 in PPD. Future randomized, placebo-controlled trials with longer follow-up periods will be essential to validate the preliminary findings presented here.
Article Information
Published Online: June 3, 2026. https://doi.org/10.4088/JCP.25m16284
© 2026 Physicians Postgraduate Press, Inc.
Submitted: December 19, 2025; accepted April 21, 2026.
To Cite: Johnson M, Aceves Baldo P, Arbe E, et al. Inhaled mebufotenin (GH001) for adult patients with postpartum depression: a phase 2a open-label clinical trial. J Clin Psychiatry 2026;87(3):25m16284.
Author Affiliations: St. Pancras Clinical Research, London, United Kingdom (Johnson, Arbe, Ratcliffe); GH Research, Dublin, Ireland (Aceves Baldo, Brennan, Doolin, Gregory, Keady, Kriger, MacIsaac, Svendsen, Valcheva); Department of Psychiatry, Amsterdam UMC, University of Amsterdam, Amsterdam, the Netherlands (Cohen, Zantvoord); Department of Psychiatry, Sheffield Health and Social Care NHS Foundation Trust, Sheffield, United Kingdom (Gann, Tully); Department of Psychiatry, University of North Carolina, Chapel Hill, North Carolina, United States (Rubinow); n.cour projects GmbH, Berlin, Germany (Terwey); Feinstein Institutes for Medical Research, Northwell Health, Manhasset, New York, United States (Deligiannidis).
Corresponding Author: Kristina M. Deligiannidis, MD, Northwell, 2000 Marcus Ave, Ste 300, New Hyde Park, NY, 11042-1069 ([email protected]).
Relevant Financial Relationships: Drs Brennan, Doolin, Gregory, Keady, MacIsaac, and Valcheva; Mr Aceves Baldo; and Ms Kriger are employees and stock option holders of GH Research. Dr Ratcliffe has been a consultant for Grünenthal, Actinogen, Takeda, GSK, GW Pharma, AstraZeneca, Camurus, Cleothena, and Ipsen. Dr Rubinow has received research funding from National Institutes of Health, Baszucki Foundation, and Sage; has been on scientific advisory boards of Sage and Sensorium; has been on clinical advisory boards of Felicity Pharma and EmbarkNeuro; and has been a consultant for Brii Biosciences, GH Research, and Aldeyra Therapeutics. Dr Svendsen is an employee of Novartis, Basel, Switzerland, and is a former employee and current shareholder of GH Research. Dr Terwey is an investor in and consultant for Aidvance and a former employee and current shareholder of GH Research. Dr Deligiannidis has been a consultant for Biogen, Brii Biosciences, Gerbera Therapeutics, GH Research, Neurocentria, Reunion Neuroscience, Lipocine, and Sage Therapeutics and a principal investigator for contracted research with DuKang Pharmaceuticals, Sage, and Woebot Health. The remaining authors have nothing to disclose.
Funding/Support: This trial was sponsored by GH Research Ireland Limited (Dublin, Ireland).
Previous Presentation: The topline results from the trial were presented as a poster at the annual congress of the American Society of Clinical Psychopharmacology; May 2025; Scottsdale, Arizona.
Acknowledgment: The authors would like to thank the patient-facing teams of the participating trial sites and the clinical trial participants and their families, without whom the trial would not have been possible.
Additional Information: Dr Arbe regretfully passed away during the development of the manuscript but remains as an honorary author.
ORCID: Emilio Arbe: https://orcid.org/0000-0003-4572-8145;
Brian Brennan: https://orcid.org/0000-0003-1869-2140;
Sem Cohen: https://orcid.org/0000-0002-9733-6146;
Kristina Deligiannidis: https://orcid.org/0000-0001-7439-2236;
Kelly Doolin: https://orcid.org/0009-0006-7089-8890;
William Gann: https://orcid.org/0000-0003-3395-8250;
Martin Johnson: https://orcid.org/0000-0002-8398-5436;
Stuart Ratcliffe: https://orcid.org/0000-0003-3181-3613;
David Rubinow: https://orcid.org/0000-0003-0815-2263;
Claus Bo Svendsen: https://orcid.org/0000-0002-1661-3464;
Theis H. Terwey: https://orcid.org/0009-0008-3020-7121;
Velichka Valcheva: https://orcid.org/0000-0002-3476-0046;
Jasper Zantvoord: https://orcid.org/0000-0002-6475-902X
Supplementary Material: Available at Psychiatrist.com.
Clinical Points
- Inhaled mebufotenin (GH001) administered as a single day individualized dosing regimen demonstrated rapid and significant reductions in depressive symptoms in patients with postpartum depression, with 100% remission by Day 8.
- GH001 was well tolerated, with no serious adverse events, no sedative effects, and same-day discharge for all patients, supporting the feasibility of this treatment approach in clinical settings.
- Preliminary breastmilk pharmacokinetic data suggest that mebufotenin and its metabolites are rapidly eliminated, supporting a treatment strategy requiring only a brief interruption of breastfeeding around dosing.
Editor’s Note: We encourage authors to submit papers for consideration as a part of our Focus on Women’s Mental Health section. Please contact Marlene P. Freeman, MD, at Psychiatrist.com/contact/freeman.
References (42)

- Stewart DE, Vigod SN. Postpartum depression: pathophysiology, treatment, and emerging therapeutics. Annu Rev Med. 2019;70:183–196. PubMed CrossRef
- Field T. Postpartum depression effects on early interactions, parenting, and safety practices: a review. Infant Behav Dev. 2010;33(1):1–6. PubMed CrossRef
- Putnick DL, Sundaram R, Bell EM, et al. Trajectories of maternal postpartum depressive symptoms. Pediatrics. 2020;146(5):e20200857. doi:10.1542/peds.2020-0857. PubMed CrossRef
- Wang L, Wu T, Anderson JL, et al. Prevalence and risk factors of maternal depression during the first three years of child rearing. J Womens Health (Larchmt). 2011;20(5):711–718. PubMed CrossRef
- Wang Z, Liu J, Shuai H, et al. Mapping global prevalence of depression among postpartum women. Transl Psychiatry. 2021;11(1):543. PubMed CrossRef
- Eastwood JG, Jalaludin BB, Kemp LA, et al. Relationship of postnatal depressive symptoms to infant temperament, maternal expectations, social support and other potential risk factors: findings from a large Australian cross-sectional study. BMC Pregnancy Childbirth. 2012;12:148. PubMed CrossRef
- Kerstis B, Aarts C, Tillman C, et al. Association between parental depressive symptoms and impaired bonding with the infant. Arch Womens Ment Health. 2016;19(1):87–94. PubMed CrossRef
- Slomian J, Honvo G, Emonts P, et al. Consequences of maternal postpartum depression: a systematic review of maternal and infant outcomes. Womens Health (Lond). 2019;15:1745506519844044. PubMed CrossRef
- Frieder A, Fersh M, Hainline R, et al. Pharmacotherapy of postpartum depression: current approaches and novel drug development. CNS Drugs. 2019;33(3):265–282. PubMed CrossRef
- Kaufman Y, Carlini SV, Deligiannidis KM. Advances in pharmacotherapy for postpartum depression: a structured review of standard-of-care antidepressants and novel neuroactive steroid antidepressants. Ther Adv Psychopharmacol. 2022;12:20451253211065859. PubMed CrossRef
- Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry. 2021;78(9):951–959. PubMed CrossRef
- Deligiannidis KM, Citrome L, Huang MY, et al. Effect of zuranolone on concurrent anxiety and insomnia symptoms in women with postpartum depression. J Clin Psychiatry. 2023;84(1):45307.
- Deligiannidis KM, Meltzer-Brody S, Maximos B, et al. Zuranolone for the treatment of postpartum depression. Am J Psychiatry. 2023;180(9):668–675. PubMed CrossRef
- Shen HW, Jiang XL, Winter JC, et al. Psychedelic 5-methoxy-N,N-dimethyltryptamine: metabolism, pharmacokinetics, drug interactions, and pharmacological actions. Curr Drug Metab. 2010;11(8):659–666. PubMed CrossRef
- Halberstadt AL, Nichols DE, Geyer MA. Behavioral effects of alpha,alpha,beta,beta-tetradeutero-5-MeO-DMT in rats: comparison with 5-MeO-DMT administered in combination with a monoamine oxidase inhibitor. Psychopharmacol Berl. 2012;221(4):709–718. PubMed CrossRef
- Reckweg J, Mason NL, van Leeuwen C, et al. A phase 1, dose-ranging study to assess safety and psychoactive effects of a vaporized 5-methoxy-N,N-dimethyltryptamine formulation (GH001) in healthy volunteers. Front Pharmacol. 2021;12:760671. PubMed CrossRef
- Reckweg JT, van Leeuwen CJ, Henquet C, et al. A phase 1/2 trial to assess safety and efficacy of a vaporized 5-methoxy-N,N-dimethyltryptamine formulation (GH001) in patients with treatment-resistant depression. Clinical trial. Front Psychiatr. 2023;14:1133414. PubMed CrossRef
- World Medical Association. Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;20:2191–2194.
- Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(Suppl 20):22–33.
- Psychedelic Drugs. Considerations for Clinical Investigations Guidance for Industry. Draft Guidance; 2023.
- Reckweg JT, Mason NL, Theunissen EL, et al. Evaluation of the peak experience scale as a rapid assessment tool for the strength of a psychoactive experience with 5-MeO-DMT. Front Psychol. 2025;16:1543640. PubMed CrossRef
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. PubMed CrossRef
- Guy W. Clinical global impressions. ECDEU Assessment Manual for Psychopharmacology. Natl Inst Ment Health Psychopharmacol Res Branch. 1976:217–222.
- Barkin JL, Wisner KL, Bromberger JT, et al. Development of the Barkin Index of Maternal Functioning. J Womens Health (Larchmt). 2010;19(12):2239–2246. PubMed CrossRef
- Kim TK, Niklewski PJ, Martin JF, et al. Enhancing a sedation score to include truly noxious stimulation: the Extended Observer’s Assessment of Alertness and Sedation (EOAA/S). Br J Anaesth. 2015;115(4):569–577. PubMed CrossRef
- Overall JE, Gorham DR. The brief psychiatric rating scale. Psychol Rep. 1962;10(3):799–812.
- Bremner JD, Krystal JH, Putnam FW, et al. Measurement of dissociative states with the Clinician-Administered Dissociative States Scale (CADSS). J Trauma Stress. 1998;11(1):125–136. PubMed CrossRef
- Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. PubMed CrossRef
- CANTAB. CANTAB the most sensitive and validated cognitive research software available. https://www.cambridgecognition.com/cantab
- Barrett FS, Bradstreet MP, Leoutsakos JS, et al. The Challenging Experience Questionnaire: characterization of challenging experiences with psilocybin mushrooms. J Psychopharmacol. 2016;30(12):1279–1295. PubMed CrossRef
- Barrett FS, Johnson MW, Griffiths RR. Validation of the revised Mystical Experience Questionnaire in experimental sessions with psilocybin. J Psychopharmacol. 2015;29(11):1182–1190. PubMed CrossRef
- Rucker JJ, Roberts C, Seynaeve M, et al. Phase 1, placebo-controlled, single ascending dose trial to evaluate the safety, pharmacokinetics and effect on altered states of consciousness of intranasal BPL-003 (5-methoxy-N,N-dimethyltryptamine benzoate) in healthy participants. J Psychopharmacol. 2024:2698811241246857.
- Vogt SB, Ley L, Erne L, et al. Acute effects of intravenous DMT in a randomized placebo-controlled study in healthy participants. Transl Psychiatry. 2023;13(1):172. PubMed CrossRef
- Strassman RJ, Qualls CR. Dose-response study of N,N-dimethyltryptamine in humans. I. Neuroendocrine, autonomic, and cardiovascular effects. Arch Gen Psychiatry. 1994;51(2):85–97. PubMed CrossRef
- Wsol A. Cardiovascular safety of psychedelic medicine: current status and future directions. Pharmacol Rep. 2023;75(6):1362–1380. PubMed CrossRef
- Kanes S, Colquhoun H, Gunduz-Bruce H, et al. Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet. 2017;390(10093):480–489. PubMed CrossRef
- Meltzer-Brody S, Colquhoun H, Riesenberg R, et al. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet. 2018;392(10152):1058–1070. PubMed CrossRef
- GH Research Announces Primary Endpoint Met in Phase 2b Trial with GH001 in TRD Demonstrating -15.5 Point Placebo-adjusted MADRS Reduction. 2025. Accessed September 17, 2025. https://investor.ghres.com/news-releases/news-release-details/gh-research-announces-primary-endpoint-met-phase-2b-trial-gh001
- GH Research Announces Primary Endpoint Met in Two Phase 2a POC Trials with GH001 and Completion of All FDA Requests to Address IND Hold with No Findings of Respiratory Toxicity in Non-Rodents. 2025. Accessed September 17, 2025. https://investor.ghres.com/news-releases/news-release-details/gh-research-announces-primary-endpoint-met-two-phase-2a-poc
- Huang D, Luo Z, Gong X, et al. Post marketing safety assessment of the novel postpartum depression drug, zuranolone: evidence from real-world pharmacovigilance analysis based on the FDA adverse event reporting system. Front Psychiatry. 2025;16:1517773. PubMed CrossRef
- Reckweg JT, Uthaug MV, Szabo A, et al. The clinical pharmacology and potential therapeutic applications of 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT). J Neurochem. 2022;162(1):128–146. PubMed CrossRef
- Clarke DE, De Faria L, Alpert JE, et al. Perinatal Mental and Substance Use Disorder: White Paper. American Psychiatric Association; 2023.
This PDF is free for all visitors!




